User login
WHAT IS AMYLOIDOSIS?
Amyloidosis is a protein deposition disease in which a specific precursor protein pathologically misfolds from its physiologic tertiary structure into a more linear shape dominated by beta-pleated sheets. The misfolded protein aggregates into oligomers, eventually forming insoluble amyloid fibrils that deposit extracellularly in tissues. Both the circulating oligomers, which are cytotoxic, and the fibrils, which cause distortion of the tissue architecture, lead to organ dysfunction. Amyloid fibrils are rigid, nonbranching structures, 7 to 10 nanometers in diameter, with a characteristic appearance on electron microscopy. Affinity for Congo red staining, which binds to the beta-pleated sheets, produces the pathognomonic “apple-green” birefringence when visualized under polarized light microscopy. Universal to all amyloid fibrils are chaperone proteins such as serum amyloid P (SAP) and glycosaminoglycans, as well as calcium. There are more than 30 different precursor proteins implicated in various amyloid diseases, arising as hereditary or nonhereditary, localized or systemic, with different organ involvement and prognosis.1–3
TWO MAIN TYPES OF CARDIAC AMYLOIDOSIS

Light chain amyloidosis (AL)
AL, formerly called primary amyloidosis, is a clonal plasma cell disorder due to the overproduction and misfolding of antibody light chain fragments. It is a rare disease with about 3,000 new cases per year in the United States.5 The median age at diagnosis is 63, although it can present in patients in their 30s and 40s.5,6 It is a systemic disease that often affects the heart, but it can affect several other organs, most commonly the kidneys, gastrointestinal (GI) tract, and nervous system.7
AL is a more aggressive disease than ATTR, with a median untreated survival of less than 6 months in patients who present with heart failure.8 Early diagnosis is crucial as mortality is high without prompt treatment.
Transthyretin amyloidosis (ATTR)
ATTR is due to misfolding of the liver-derived precursor protein transthyretin (TTR) (previously called prealbumin), either as an acquired wild-type variant (ATTRwt) or as a hereditary mutant variant (ATTRm). ATTRwt, known previously as senile CA, typically affects older males and presents as a late onset hypertrophic restrictive cardiomyopathy, often preceded by carpal tunnel syndrome or spinal stenosis or both. The ATTRm variant, caused by one of many different point mutations in the TTR gene, can manifest as a polyneuropathy, cardiomyopathy, or a mixed phenotype that varies according to the specific mutation.
While ATTR portends a better prognosis than AL, it is still a progressive disorder with significantly reduced survival and quality of life. The median survival of patients with the ATTRwt variant is about 4 years and for patients with the ATTRm variant, survival depends on the mutation.9 TTR is a protein tetramer composed of 4 identical 127-amino acid monomers noncovalently bound at a dimer-dimer interface (Figure 1). It is a transport protein for thyroxine and retinol binding protein. The dissociation of the tetramer is the rate-limiting step for amyloid fibrillogenesis. Differentiating the ATTRwt variant and the ATTRm variant is done by testing the TTR gene for a mutation.1,3
How common is the ATTRwt variant? The ATTRwt variant is often an unrecognized cause of diastolic heart failure in the elderly, with up to 25% of patients 85 and older showing ATTRwt amyloid deposits on autopsy studies.10 A recent study showed that 13% of patients 60 and older hospitalized with heart failure with preserved ejection fraction had grade 2 to 3 uptake on 99mtechnetium-pyrophosphate (99mTcPYP) scintigraphy, which is consistent with ATTR-CA.11 In 43 consecutive patients undergoing transcatheter aortic valve replacement, 11.6% were found to have significant uptake on 99mTcPYP scan.12 It is clear given the aging population that the ATTRwt variant will become the most common form of amyloidosis. It is much more common in white males, with a median age at diagnosis of 75.13 Carpal tunnel syndrome (almost always bilateral) and spinal stenosis are present in about 50% of patients diagnosed with ATTRwt-CA and often precede clinical presentation of heart failure by 5 to 15 years.14–17
How common is the ATTRm variant? There are more than 100 point mutations in the TTR gene that lead to various familial TTR-related amyloid syndromes, either neuropathic (familial amyloid polyneuropathy [FAP]) or cardiomyopathic (familial amyloid cardiomyopathy).18 The most common mutation in the United States is V122I in which there is an isoleucine substitution for valine at the 122nd amino acid position. This mutation is seen in African Americans, 3% to 4% of whom are heterozygote carriers.19 Although the true penetrance is unknown, this mutation can lead to a late-onset restrictive cardiomyopathy with minimal neuropathy and is frequently misdiagnosed as hypertensive heart disease or diastolic heart failure. The median survival for V122I ATTRm-CA is about 2 years but likely depends on the stage at the time of diagnosis.20 The second most common mutation in the United States, T60A, is seen in patients of Irish descent and causes a mixed neuropathy and cardiomyopathy.17
PATHOLOGY AND PATHOPHYSIOLOGY OF CA

The atria are universally involved with interatrial septal thickening, which can lead to poor atrial function and increased rates of atrial fibrillation (ATTR more so than AL).21,29 The conduction system can be affected causing varying degrees of heart block, as well as bundle branch block (ATTR more so than AL).30 The valves are usually thickened, often associated with mild to moderate regurgitation. Pericardial involvement can lead to small pericardial effusions (large effusions are rare), and coronary involvement (classically, small intramural vessels) can lead to ischemia and angina with normal epicardial coronaries (AL more so than ATTR).31–33
Thickened left and right ventricular walls result in a nondilated ventricle that is stiff and poorly compliant, resulting in progressive diastolic filling abnormalities. Systolic dysfunction can be seen in severe and advanced disease. Importantly, ejection fraction measured by echocardiography is misleading in CA, as reduced end-diastolic volume produces a low stroke volume. For example, an ejection fraction of 50%, when starting at a significantly reduced end-diastolic volume (for example, 70 mL), leads to a significantly reduced stroke volume (35 mL) and, thus, cardiac output. This explains why patients with CA cannot usually tolerate reduced heart rates, as their cardiac output is dependent on heart rate.34–36
CLINICAL PRESENTATION
Patients with CA typically exhibit heart failure with preserved ejection fraction (otherwise known as diastolic heart failure). Dyspnea on exertion is common; however, some patients can present with more right-sided heart failure symptoms such as lower-extremity edema and ascites. Fatigue and weakness are related to low cardiac output and often attributed to nonspecific symptoms of aging. Because of the thickened ventricles, patients can often be misdiagnosed as having HCM with or without obstruction.7,34 The first manifestation of CA may be atrial fibrillation, most commonly in ATTRwt-CA, or cardioembolic stroke. Atrial fibrillation can be present for years before CA is considered. Bundle branch block and complete heart block (more common in ATTR-CA than AL-CA) may lead to pacemaker implantation.30 Angina with normal coronaries can occur, and a rare presentation may be cardiogenic shock due to diffuse ischemia.31–33 Elderly patients with CA can present with low-flow, low-gradient aortic stenosis.37

DIAGNOSIS

ECG
As opposed to that seen in true left ventricular hypertrophy (LVH), which leads to increased voltage on ECG, amyloid infiltration of the myocardium leads to lower voltage. Thus, what is indicative of LVH on echocardiogram combined with low voltage on the ECG is a classic finding for CA. However, only about 50% of patients with AL-CA and about 30% of patients with ATTR-CA meet true low-voltage criteria (QRS amplitude less than 5 mm in limb leads or less than 10 mm in precordial leads).30,38 Hence, the absence of low-voltage criteria does not exclude the diagnosis of CA. Approximately 10% of patients with CA confirmed by biopsy met ECG criteria for LVH.38 The key point is to consider the overall degree of voltage on the ECG relative to the degree of LV thickening on the echocardiogram, recognizing that lower voltage than what would be expected may indicate possible infiltrative disease such as CA. The other main finding on the ECG in patients with CA is a pseudoinfarct pattern with Q waves in the early precordial leads mimicking a prior anteroseptal myocardial infarction.38,39 This finding is seen in about 50% of patients (Figure 3).39 Wide QRS complexes are more frequent in ATTR-CA and lower limb voltages are more frequent in AL-CA.30
Echocardiogram
The echocardiographic finding of LVH in patients with CA is misleading in that the LV thickening is due to infiltrating amyloid fibrils and not to myocyte hypertrophy. That said, the terms LVH and LV thickening are used interchangeably when describing the echocardiographic phenotype. LV wall thickness greater than 12 mm (6 mm to 10 mm is normal) in the absence of hypertension should prompt suspicion for CA.34 LV thickening most often appears symmetric; however, occasionally it may exhibit asymmetric septal hypertrophy, particularly in ATTRwt-CA. In some cases, there may be a smaller subset that can actually have dynamic LV outflow obstruction similar to that seen in hypertrophic obstructive cardiomyopathy.22–24 An important echocardiographic clue that can differentiate CA from other diseases is thickening of both the LV and right ventricle (Figure 3). Septal wall thickness and LV mass index are greater in ATTR-CA compared with AL-CA.30 On average, the LV septum is around 15 mm in AL-CA and around 18 mm in ATTRwt-CA.40 Historically, the characteristic myocardial “granular sparking” or “speckling” pattern has low sensitivity and specificity.16 The left ventricle is not dilated; rather, the ventricular dimensions are usually smaller than normal. Although ejection fraction is usually preserved, cardiac output is low due to decreased ventricular volume.37 Systolic dysfunction occurs late in the disease.16 Diastolic dysfunction is universal, with a mitral inflow pattern that can range from stage I (abnormal relaxation) in early disease to stage III (restrictive filling pattern) in more advanced disease. Septal and lateral tissue Doppler velocities are very low in amyloid heart disease.41 Another echocardiographic clue to diagnosis is thickening of the heart valves, which is not seen in hypertensive heart disease or hypertrophic cardiomyopathy. Biatrial dilation is common, and there can be thickening of the interatrial septum.
Laboratory testing
N-terminal pro-b-type natriuretic peptide (NT-proBNP) is universally elevated in CA and is typically higher in AL-CA than in ATTR-CA. Troponin T or troponin I or both may be chronically elevated in CA and likely signify small-vessel ischemia. In the appropriate clinical context of a thickened ventricle and heart failure, an elevated troponin value (outside of an acute coronary syndrome) should trigger suspicion for CA. Workup for a monoclonal protein process should always be done when considering CA to rule out AL. Serum and urine protein electrophoresis are insensitive tests to detect AL and should not be relied upon as a screening test. The serum free light chain (sFLC) assay, which measures free kappa and lambda light chain levels and reports the ratio, is a sensitive test that should be measured routinely along with immunofixation of the serum and urine. In AL, sFLC will reveal an abnormal kappa-lambda ratio. An abnormally low ratio (less than 0.26) suggests a monoclonal lambda light chain process, while an abnormally high ratio (greater than 1.65) suggests a monoclonal kappa light chain process. Immunofixation will reveal an M-protein. Because light chains are excreted by the kidney, the serum levels of both kappa and lambda will be elevated in renal dysfunction, but the ratio should remain normal.16,31,34,40,42–47
Advanced noninvasive diagnostic tools for CA
Over the past decade, the ability to diagnose CA noninvasively has dramatically improved with strain imaging using 2D speckle tracking echocardiography, cardiac magnetic resonance imaging (MRI), and nuclear bone scintigraphy. These diagnostic tools have given clinicians options to pursue the diagnosis of CA without directly proceeding to endomyocardial biopsy.

Cardiac MRI. Cardiac MRI is useful for the diagnosis of CA (Figure 4B, 4C). Imaging after administration of gadolinium contrast shows a characteristic late gadolinium enhancement (LGE) pattern that is diffuse and subendocardial, and does not follow any particular coronary distribution.49 LGE can also be seen in the right ventricle and the atrial walls, and can be transmural and patchy in ATTRwt-CA. This pattern is highly sensitive (93%) and specific (70%) for CA with an overall negative predictive accuracy of 84%.50
One of the main limitations of cardiac MRI for the diagnosis of CA is the inability to give contrast in patients with reduced glomerular filtration rate. However, native T1-myocardial mapping techniques that do not require contrast show significantly increased native T1 times in CA and offer a promising alternative. Cardiac MRI parameters such as LGE, the difference in inversion time between the LV cavity and myocardium, native T1 mapping, and extracellular volume offer prognostic information. A greater than fivefold mortality increase is seen in CA patients with transmural LGE compared with those without LGE.49,50
99mTcPYP scintigraphy. 99mTcPYP, a radiotracer used in bone scans, was initially used in cardiology to quantify myocardial infarction due to its ability to localize calcium.51 Its potential utility in CA came in 1982 when diffuse myocardial 99mTcPYP uptake on cardiac radionucleotide imaging was noted in 10 patients with tissue-proven amyloidosis.52 Several subsequent studies reproduced and expanded upon this observation and revealed its diagnostic value, specifically showing that there is significant uptake in ATTR-CA and no to mild uptake in AL-CA. This offers a significant advantage over other noninvasive modalities in that it not only confirms the diagnosis of CA but differentiates ATTR-CA.16
99mTcPYP myocardial radiotracer uptake is graded by the semiquantitative visual score of cardiac retention, where grade 0 = no cardiac uptake, grade 1 = mild uptake less than bone, grade 2 = moderate uptake equal to bone, and grade 3 = high uptake greater than bone (Figure 4D).42 Additionally, quantitative analysis of heart retention can be calculated drawing circular regions of interest over the heart and mirrored on the contralateral chest wall. A heart-to-contralateral ratio greater than 1.5 is consistent with the diagnosis of ATTR-CA.53,54 In 2016, a multicenter study showed that grade 2 or 3 myocardial radiotracer uptake on bone scintigraphy in the absence of evidence of a monoclonal gammopathy was diagnostic for ATTR-CA, providing a cost-effective and non-invasive technique with a specificity and positive predictive value of 100% (confidence interval, 99.0–100%).42
Endomyocardial biopsy, right heart catheterization, and fat biopsy
Endomyocardial biopsy is essentially 100% sensitive for the diagnosis of CA.25 The main risk of pursuing endomyocardial biopsy is about a 1% risk of right ventricular perforation leading to cardiac tamponade.55 The other limitation to this approach is that not all centers are equipped to perform this procedure. Birefringence under polarized light microscopy is histopathologically diagnostic of CA; however, further subtyping by the pathologist to determine if it is AL or ATTR is absolutely crucial. Subtyping can be performed by immunohistochemistry with caution taken for misinterpretation. If there is any question of accuracy, the specimen should be sent for laser microdissection and mass spectroscopy for accurate identification of the precursor protein type (some centers routinely perform mass spectroscopy on all myocardial specimens).16,31
Right heart catheterization is nonspecific and shows restrictive hemodynamics. The right atrial waveform shows rapid x and y descents and the right ventriclular tracing may show a dip-and-plateau pattern typical of restrictive cardiomyopathy. Cardiac output can be preserved but more commonly is low.16,31
Fat pad biopsy is 60% to 80% sensitive in AL, 65% to 85% sensitive in ATTRm, and only 14% sensitive in ATTRwt, with the accuracy dependent on the operator, pathologist, and how much tissue is removed (fat pad aspirate vs biopsy specimen).56–58 Fat pad biopsy has diagnostic limitations, and a negative fat pad biopsy does not rule out amyloidosis.
DIAGNOSTIC ALGORITHM FOR CARDIAC AMYLOIDOSIS
.")
In conjunction with laboratory tests, 99mTcPYP scan of the heart can be ordered to investigate the possibility of ATTR-CA. Grade 2 to 3 myocardial uptake in the absence of a monoclonal plasma cell process is consistent with the diagnosis of ATTR-CA. Grade 0 or 1 myocardial uptake on 99mTcPYP scan with an abnormal sFLC ratio or positive M protein on immunofixation suggests AL-CA and a bone marrow biopsy should be performed. If the patient has an abnormal sFLC ratio and grade 2 to 3 uptake on 99mTcPYP scan, the diagnosis of ATTR-CA with unrelated monoclonal gammopathy of undetermined significance should be considered. However, this would need to be reconciled by pursuing endomyocardial biopsy and accurate tissue typing. If the 99mTcPYP scan is negative, and the sFLC ratio is normal, and immunofixation is negative, a diagnosis of CA is very unlikely.16,31,40,42–47
If the diagnosis of ATTR-CA is made, genetic testing can determine the presence or absence of a mutation to differentiate ATTRm or ATTRwt, respectively. If the diagnosis of AL-CA is suggested, a bone marrow biopsy is necessary to identify and quantify the plasma cell clone.16,31,40,42–47
TREATMENT

Management of heart failure in cardiac amyloidosis
The main treatment of heart failure revolves around sodium restriction and diuretics to relieve congestion. This can prove challenging in many patients due to the narrow window between too high or too low filling pressures. A combination of loop diuretics and an aldosterone antagonist is most effective.31,34,44,45 Torsemide is preferred over furosemide due to its superior bioavailability and longer duration of action, particularly since these patients have issues with gut edema and GI absorption. Due to dependence of the cardiac output on heart rate and the tendency for orthostatic hypotension, traditional neurohormonal antagonists including beta blockers and angiotensin-converting enzyme inhibitors are neither effective nor well tolerated.60 However, in patients with atrial fibrillation, beta blockers may need to be used for rate control. Nondihydropyridine calcium channel blockers bind avidly to amyloid fibrils and are contraindicated due to risk of profound hypotension and syncope.30,31,34,44,45 Digoxin is usually avoided in CA due to concerns of increased risk of toxicity; however, it may be used with caution for rate control in atrial fibrillation given its lack of negative inotropy.61 Maintenance of normal sinus rhythm is preferable due to the importance of atrial contribution to cardiac output.
Anticoagulation in patients with atrial fibrillation and even in patients with normal sinus rhythm and poor atrial function is important due to the high risk of thromboembolic complications.62 Pacemakers are indicated for heart block or symptomatic bradycardia.63 The role of intracardiac defibrillators is controversial, but may be warranted in selected patients with AL-CA.64,65
AL treatment
Risk stratification and prognostication for AL. The most important determinant of clinical outcome in AL is the extent of cardiac involvement, as congestive heart failure and sudden cardiac death are the most common causes of death. The level of NT-proBNP and the level of either troponin T or troponin I have strong prognostic value and form the basis for the staging system in AL. Various iterations have evolved over the years, but the most widely adopted is the 4-stage system developed and validated by Mayo Clinic. This system uses a cutoff value at diagnosis for NT-proBNP greater than 1,800 ng/mL, troponin T greater than 0.025 μg/L, and the difference between kappa and lambda free light chain levels greater than 180 mg/L. Stage level increases by the number of cutoff values exceeded, with stage IV carrying a median survival of 6 months.47 Additionally, the troponin T level can help risk-stratify patients being considered for autologous stem cell transplant. In a retrospective study, troponin T greater than 0.06 μg/L was associated with increased mortality following stem cell transplant.66 Ultimately, prognosis in AL-CA is related to the hematologic response to chemotherapy.
Current treatment strategies for AL. The survival of patients with AL has improved over the years with the advent of more effective chemotherapeutic regimens that kill the underlying plasma cell clone producing the unstable light chains. The goal of treatment is to achieve a complete hematologic response with normalization of the affected light chain and sFLC ratio as well as elimination of the M protein on immunofixation.
The development of the proteasome inhibitor bortezomib has improved efficacy and survival in AL causing a faster and more complete hematologic response than prior regimens.43,67,68
The most commonly used first-line treatment consists of a 3-drug combination with the alkylating agent cyclophosphamide, the proteasome inhibitor bortezomib, and the steroid dexamethasone, which is given weekly.43 A retrospective study by Sperry et al8 showed that patients receiving an alkylating agent, bortezomib, and a steroid had the best outcomes compared with other regimens.
For patients with refractory or relapsed disease, the CD38 monoclonal antibody daratumumab can be used if patients meet myeloma criteria and has been found to be effective thus far.68,69 Newer proteasome inhibitors such as ixazomib, which is taken orally, are being studied in alternative combination regimens.70 High-dose chemotherapy with autologous stem cell transplant can be considered in patients with an acceptable cardiac risk profile and may offer more complete and durable remission than chemotherapy, although this is controversial.43,71
The cardiologist’s role in AL-CA. The hematologist directs the chemotherapy for AL but works closely with the cardiologist when there is cardiac involvement. The main role of the cardiologist is to manage volume status with diuretics, monitor for arrhythmia, and evaluate the cardiac response to treatment.43,71 Cardiac response was traditionally measured by echocardiographic changes of wall thickness, diastolic function, and ejection fraction, as well as changes in New York Heart Association (NYHA) functional class.67 However, it is uncommon to see reduction in LV wall thickness or significant improvement in ejection fraction and if it does occur, it is a slow process that usually takes more than 1 year. With the advent of longitudinal myocardial strain imaging, improvements in strain can be seen despite the lack of structural changes on echocardiogram.72 In 2012, consensus criteria defined a cardiac response as a greater than 30% reduction in NT-proBNP.73 Misfolded light chains are toxic to cardiomyocytes by causing increased oxidative stress and impairing contractility. Thus, reduction in light chain levels can lead to clinical improvement and significant reductions in NT-proBNP without changing amyloid fibril burden in the heart.
Heart transplant for patients with AL-CA. For patients who have a good hematologic response to initial chemotherapy but have limited predicted survival due to severe heart failure, heart transplant followed by autologous stem cell transplant, is a treatment strategy that can be considered. The patient must have clinically isolated severe cardiac disease, minimal amyloid burden in other organs, and a plasma cell clone that is responsive to therapy. Initial reports of heart transplant showed poor survival rates due to recurrent amyloid in the transplanted heart and progressive amyloid deposition in other organs. However, due to improved anti-plasma-cell-directed therapy and refinement in patient selection, outcomes have improved. Contemporary series of patients undergoing heart transplant followed by stem cell transplant showed that outcomes are almost comparable to heart transplant for other indications, with a 5-year survival rate of approximately 65%.74
Future therapies for AL. There is an AL amyloid-directed monoclonal antibody designed to remove amyloid fibrils from affected organs and is currently undergoing clinical trials. NEOD001, a humanized murine monoclonal antibody that targets an epitope exposed during light chain misfolding, binds to the light chain amyloid fibril and signals an immune response to clear the deposits. This agent has completed phase 1 and 2 clinical trials of 27 patients previously treated with at least 1 plasma cell-directed therapy. It showed good tolerability and achieved both renal and cardiac responses in most patients.75
A phase 2b clinical trial (NCT02632786) of patients with AL with a previous hematologic response to treatment and persistent heart dysfunction is underway and expected to be completed in January 2018. A phase 3 clinical trial (NCT02312206) of NEOD001 as an adjunct to chemotherapy is also ongoing and results are expected in February 2019.
ATTR treatment
Liver transplant for ATTRm amyloidosis for the V30M mutation that causes FAP was first described in 1990, but it has not been well validated in other mutations and is not a solution for ATTRwt.76 Significant progress has been made over the past 2 decades in the understanding of the pathophysiologyy of ATTR, paving the way for promising advancements in pharmacotherapy. Presently, there are 3 classes of pharmacologic agents, grouped by the point of disease process each strategy targets:
- Block TTR synthesis at the translational level in hepatocytes
- Stabilize the TTR tetramer to inhibit the rate-determining step of amyloidogenesis
- Disrupt and clear the ATTR amyloid fibril.77
Block TTR synthesis. TTR messenger RNA (mRNA) can be targeted by “silencers” preventing translation, thereby reducing the production TTR protein by hepatocytes. The resultant sustained reduction of plasma TTR should decrease or halt amyloid deposition by making less TTR available to dissociate and deposit in the heart and nerves. There are 2 approaches to silencing TTR mRNA translation: small interfering RNA (siRNA) and antisense oligonucleotide (ASO).77
An siRNA, packaged in a lipid nanoparticle to ensure delivery to the liver, has been designed to bind to a conserved region of TTR mRNA, degrading the mRNA and reducing TTR protein expression. One siRNA, patisiran, completed the phase 3 APOLLO clinical trial (NCT01960348) in August 2017. It is an intravenous medication that requires premedication. The APOLLO trial studied patients with neuropathic variants of ATTRm, with a primary end point of neuropathy progression over 18 months. Patisrian met the primary end point and will likely be approved by the US Food and Drug Administration (FDA) by the third quarter of 2018. Given its target of a conserved 3’ untranslated region of TTR mRNA, patisiran should theoretically yield benefits not just in FAP but to both ATTRm-CA and ATTRwt-CA.68,77
ASOs are single-stranded oligonucleotides, typically 20 nucleotides in length, that bind to mRNA and elicit enzymatic mRNA degradation and reduced protein expression. Inotersen (IONIS-TTRRx) is an ASO drug that targets a conserved region of TTR mRNA. Inotersen is administered by weekly subcutaneous injection. A phase 2/3 clinical trial (NCT01737398) completed in October 2017 studied the drug's efficacy in treating FAP, with a primary end point of neuropathy progression over 65 weeks. It met the primary end point and a subset of patients actually improved. Like patisiran, inotersen is likely to be approved by the FDA for a neuropathy indication by the third quarter of 2018 and may benefit patients with ATTRm-CA and ATTRwt-CA. More studies are needed in the ATTR cardiac population.68,77
Stabilize the TTR tetramer. The TTR tetramer has 2 thyroxine binding pockets that stabilize the structure when bound preventing dissociation. Dissociation of the tetramer, the rate limiting step for ATTR fibrillogenesis, can be reduced using pharmacologic agents that bind to the thyroxine binding pockets. Both new and repurposed agents have been found to stabilize the TTR tetramer, including diflunisal, tafamidis, tolcapone, and AG10.68,77
Diflunisal (Dolobid) is a nonacetylated salicylate nonsteroidal anti-inflammatory drug used for over 3 decades to treat arthritis and musculoskeletal pain. Unrelated to its anti-inflammatory properties, it interacts with TTR’s thyroxine binding pocket to increase the stability of the tetramer. A 2013 randomized, placebo-controlled, international multicenter trial of 130 patients with FAP demonstrated a statistically significant slowing of polyneuropathy progression; however, 67 patients (diflunisal n = 27, placebo n = 40) did not complete the study.78 A small study revealed diflunisal to be reasonably well-tolerated in ATTR-CA, and to date it is the only readily available pharmacotherapy for ATTR supported by a randomized, placebo-controlled trial.79,80 Diflunisal may be considered for off-label use in patients with ATTR-CA with relatively preserved kidney function and no increased bleeding risk, taken under the supervision of a cardiologist who will monitor for fluid retention and changes in renal function.77
Tafamidis (Vyndaqel), like diflunisal, interacts with TTR’s thyroxine binding pocket and increases tetrameric stability. A phase 2 open-label clinical trial studied the efficacy and tolerance of tafamidis in 31 patients with ATTRwt and NYHA functional class 1 and 2 followed for 1 year. Twenty-eight patients completed the study and 2 patients died. TTR stabilization at 6 weeks was achieved in 30 of 31 patients (96.8%), and success at 1 year in 25 of 28 patients (89.3%). No clinical progression occurred in 16 of 31 patients (51.5%), and tafamidis was generally well-tolerated, with diarrhea the most common side effect in 7 of 31 patients (22.6% ).81,82
A phase 3 clinical trial (NCT01994889) is studying tafamidis vs placebo in patients with ATTRm-CA or ATTRwt-CA (excluding NYHA functional class 4) with the primary outcome measure of all-cause mortality and heart failure-related hospitalizations over 30 months. This is a large trial that enrolled 446 patients and data collection is expected to be completed in February 2018.
Tolcapone, approved by the FDA for Parkinson disease, has been found to be a potent TTR stabilizer by binding both thyroxine binding pockets of the TTR tetramer simultaneously. However, tolcapone has an FDA “black box” warning due to the risk of potentially fatal acute fulminant liver failure and is not currently used in ATTR therapy.68 Nonetheless, it will likely undergo further study for ATTR.
Recruiting is underway for a phase 1 clinical trial of AG10, a potent selective TTR stabilizer (NCT03294707).68
Disruption and clearance of the ATTR amyloid fibril. Even though treatment directed at blocking TTR synthesis or stabilizing the tetramer may be effective at preventing further deposition, the residual amyloid deposits persist and continue to affect organ function. With that need in mind, several agents that disrupt the amyloid formation process further downstream have been evaluated at a basic science level and in a few small nonrandomized open-label studies.77
Doxycycline is a tetracycline antibiotic with demonstrated effectiveness in disrupting mature amyloid fibrils in mouse models.83 Tauroursodeoxycholic acid is a bile acid with the ability to disrupt prefibrillar amyloid components. This combination has been studied in patients with ATTR-CA in 2 small open-label trials, with only 1 having results published. There was no progression in NT-proBNP or wall thickness in a cohort of 7 patients who completed 12 months of treatment.84 These data are nonrandomized and hypothesis-generating, as adequate studies would need to be performed to evaluate this hypothesis further. Because there are currently no FDA-approved therapies for ATTR, patients may be offered this combination fully informed that the data are limited. (Note: ursodiol is substituted for tauroursodeoxycholic acid, which is not available in the United States.)68
Green tea extract contains the polyphenol epigallocatechin-3-gallate, which has shown the ability for fibril disruption as well as TTR stabilization, importantly using a binding site separate from the thyroxine binding pocket (utilized in diflunisal and tafamidis). A small open-label study of 19 patients with ATTR-CA of whom 14 took green tea extract for 1 year reported a reduction in intraventricular septal wall thickness at 1 year, and in 9 patients a reduction of 12.5% in LV mass measured by cardiac MRI.85
Curcumin, the active ingredient in the household spice turmeric, has displayed in vitro promise as a TTR stabilizer by binding to the thyroxine binding pocket, and as an amyloid fibril disruptor by increasing macrophage degradation activity. Although there have only been preliminary animal studies, this supplement may be promising for further study in humans with ATTR-CA.68
PRX004 is a synthetic antibody designed to bind to non-native misfolded forms of TTR with the goal of potentially preventing deposition and promoting clearance of TTR aggregates.86 A phase 1 open-label escalation trial (NCT03336580) in 36 patients with ATTRm is planned and, hopefully, will pave the way for further study.
Heart transplant for patients with ATTR-CA. Patients with ATTRwt-CA who are young enough to undergo heart transplant have displayed favorable outcomes given that it causes clinically isolated heart disease and is an indolent process that should not affect the transplanted heart over the average life span of the allograft. Patients with the ATTRm mutation V122I have been treated with heart transplant alone, with the thought process that again, due to the indolent nature of amyloid deposition, concomitant liver transplant may not be needed.74 Thus far, 6 patients with this mutation have undergone successful transplant at our institution with heart alone, 1 of whom is 9 years posttransplant without any recurrent amyloid in the allograft. Patients with the T60A mutation that causes both polyneuropathy and cardiomyopathy require combined heart and liver transplant.74
Are there other therapies being studied to clear amyloid deposits and reverse organ dysfunction?
Extracellular deposits of amyloid fibrils, regardless of precursor protein, contain common elements such as calcium, glycosaminoglycans, and an SAP component. SAP stabilizes amyloid fibrils and makes them resistant to degradation. A monoclonal immunoglobulin G1 anti-SAP antibody has been designed to target the ubiquitous SAP component, signaling an immune response that leads to macrophage mediated clearance of amyloid fibrils, regardless of the type. Treatment with this approach was studied in a pilot trial of 16 patients mostly with AL, and within a 6-week period some of the patients had dramatic reversal of liver amyloid deposition.2 This has paved the way for a phase 2 open-label trial to be performed in patients with AL-CA and ATTR-CA (NCT03044353) and plans for a randomized phase 3 trial in CA are in discussion. There is optimism that this therapy may achieve the holy grail of removing amyloid rather than just preventing further deposition.
CONCLUSION
The diagnosis of CA requires a high index of suspicion. The diagnostic tools have improved due to the availability of modern imaging techniques, and the advent of measuring the sFLC assay along with immunofixation of the serum and urine. The prognosis for patients with AL-CA used to be dismal, with very poor survival rates. The current treatment strategies that include proteasome inhibitors have significantly improved survival, emphasizing the importance of early diagnosis and prompt initiation of therapy. Monoclonal antibodies against plasma cells (daratumumab) and light chain amyloid deposits (NEOD001) have the potential to further improve outcomes. The diagnosis of ATTR-CA used to be a futile academic pursuit given the lack of available therapies. However, there are several new FDA-approved agents on the horizon, including TTR gene silencers and stabilizers. CA is no longer considered to be rare and hopeless. Rather, it is more common than previously recognized and even more treatable.
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med 2003; 349:583–596.
- Richards DB, Cookson LM, Berges AC, et al. Therapeutic clearance of amyloid by antibodies to serum amyloid P component. N Engl J Med 2015; 373:1106–1114.
- Sipe JD, Benson MD, Buxbaum JN, et al. Nomenclature 2014: amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 2014; 21:221–224.
- Maleszewski JJ. Cardiac amyloidosis: pathology, nomenclature, and typing. Cardiovasc Pathol 2015; 24:343–350.
- Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood 1992; 79:1817–1822.
- Krsnik I, Cabero M, Morillo D, et al. Light chain amyloidosis: Experience in a tertiary hospital: 2005-2013. Rev Clin Esp 2015; 215:1–8.
- Dubrey SW, Cha K, Anderson J, et al. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM 1998; 91:141–157.
- Sperry BW, Ikram A, Hachamovitch R, et al. Efficacy of chemotherapy for light-chain amyloidosis in patients presenting with symptomatic heart failure. J Am Coll Cardiol 2016; 67:2941–2948.
- Gillmore JD, Damy T, Fontana M, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J 2017; Oct 18. [Epub ahead of print] doi:10.1093/eurheartj/ehx589.
- Tanskanen M, Peuralinna T, Polvikoski T, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med 2008; 40:232–239.
- González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015; 36:2585–2594.
- Longhi S, Lorenzini M, Gagliardi C, et al. Coexistence of degenerative aortic stenosis and wild-type transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging 2016;9:325–327.
- Grogan M, Scott CG, Kyle RA, et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol 2016; 68:1014–1020.
- Gioeva Z, Urban P, Meliss RR, et al. ATTR amyloid in the carpal tunnel ligament is frequently of wildtype transthyretin origin. Amyloid 2013; 20:1–6.
- Yanagisawa A, Ueda M, Sueyoshi T, et al. Amyloid deposits derived from transthyretin in the ligamentum flavum as related to lumbar spinal canal stenosis. Mod Pathol 2015; 28:201–207.
- Siddiqi OK, Ruberg FL. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med 2017; Jul 13. [Epub ahead of print] doi:10.1016/j.tcm.2017.07.004.
- Nakagawa M, Sekijima Y, Yazaki M, et al. Carpal tunnel syndrome: A common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid 2016; 23:58–63.
- Sekijima Y, Yoshida K, Tokuda T, Ikeda S-I. Familial Transthyretin Amyloidosis. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. Seattle, WA: University of Washington, Seattle; 1993–2017. Available at: https://www-ncbi-nlm-nih-gov.ccmain.ohionet.org/books/NBK1194. Updated January 26, 2012. Accessed November 16, 2017.
- Buxbaum J, Jacobson DR, Tagoe C, et al. Transthyretin V122I in African Americans with congestive heart failure. J Am Coll Cardiol 2006; 47:1724–1725.
- Ruberg FL, Maurer MS, Judge DP, et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: The Transthyretin Amyloidosis Cardiac Study (TRACS). Am Heart J 2012; 164:222–228.e1.
- Wright JR, Calkins E. Amyloid in the aged heart: frequency and clinical significance. J Am Geriatr Soc 1975; 23:97–103.
- Vermeer AMC, Janssen A, Boorsma PC, Mannens MMAM, Wilde AAM, Christiaans I. Transthyretin amyloidosis: a phenocopy of hypertrophic cardiomyopathy. Amyloid 2017; 24:87–91.
- Velazquez-Ceceña JL, Lubell DL, Nagajothi N, Al-Masri H, Siddiqui M, Khosla S. Syncope cardiomyopathy in a patient with primary AL-type amyloid heart disease. Tex Heart Inst J 2009; 36:50–54.
- Stegman BM, Kwon D, Rodriguez ER, Hanna M, Cho L. Left ventricular hypertrophy in a runner: things are not always what they seem. Circulation 2014; 130:590–592.
- Ardehali H, Qasim A, Cappola T, et al. Endomyocardial biopsy plays a role in diagnosing patients with unexplained cardiomyopathy. Am Heart J 2004; 147:919–923.
- Berghoff M, Kathpal M, Khan F, Skinner M, Falk R, Freeman R. Endothelial dysfunction precedes C-fiber abnormalities in primary (AL) amyloidosis. Ann Neurol 2003; 53:725–730.
- Khan MF, Falk RH. Amyloidosis. Postgrad Med J 2001; 77:686–693.
- Martinez-Naharro A, Treibel TA, Abdel-Gadir A, et al. Magnetic resonance in transthyretin cardiac amyloidosis. J Am Coll Cardiol 2017; 70:466–477.
- Longhi S, Quarta CC, Milandri A, et al. Atrial fibrillation in amyloidotic cardiomyopathy: prevalence, incidence, risk factors and prognostic role. Amyloid 2015; 22:147–155.
- Sperry BW, Vranian MN, Hachamovitch R, et al. Are classic predictors of voltage valid in cardiac amyloidosis? A contemporary analysis of electrocardiographic findings. Int J Cardiol 2016; 214:477–481.
- Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation 2005; 112:2047–2060.
- Berk JL, Keane J, Seldin DC, et al. Persistent pleural effusions in primary systemic amyloidosis: etiology and prognosis. Chest 2003; 124:969–977.
- Miani D, Rocco M, Alberti E, Spedicato L, Fioretti PM. Amyloidosis of epicardial and intramural coronary arteries as an unusual cause of myocardial infarction and refractory angina pectoris. Ital Heart J 2002; 3:479–482.
- Mesquita ET, Jorge AJL, Souza CV Junior, Andrade TR. Cardiac amyloidosis and its new clinical phenotype: heart failure with preserved ejection fraction. Arg Bras Cardiol 2017; 109:71–80.
- Swanton RH, Brooksby IAB, Davies MJ, Coltart DJ, Jenkins BS, Webb-Peploe MM. Systolic and diastolic ventricular function in cardiac amyloidosis: studies in six cases diagnosed with endomyocardial biopsy. Am J Cardiol 1977; 39:658–664.
- Tyberg TI, Goodyer AVN, Hurst VW 3rd, Alexander J, Langou RA. Left ventricular filling in differentiating restrictive amyloid cardiomyopathy and constrictive pericarditis. Am J Cardiol 1981; 47:791–796.
- Sperry BW, Jones BM, Vranian MN, Hanna M, Jaber WA. Recognizing transthyretin cardiac amyloidosis in patients with aortic stenosis: impact on prognosis. JACC Cardiovasc Imaging 2016; 9:904–906.
- Cyrille NB, Goldsmith J, Alvarez J, Maurer MS. Prevalence and prognostic significance of low QRS voltage among the three main types of cardiac amyloidosis. Am J Cardiol 2014; 114:1089–1093.
- Hongo M, Yamamoto H, Kohda T, et al. Comparison of electrocardiographic findings in patients with AL (primary) amyloidosis and in familial amyloid polyneuropathy and anginal pain and their relation to histopathologic findings. Am J Cardiol 2000; 85:849–853.
- Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation 2017; 135:1357–1377.
- Abdalla I, Murray RD, Lee JC, Stewart WJ, Tajik AJ, Klein AL. Duration of pulmonary venous atrial reversal flow velocity and mitral inflow a wave: new measure of severity of cardiac amyloidosis. J Am Soc Echocardiogr 1998; 11:1125–1133.
- Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016; 133:2404–2412.
- Gertz MA. Immunoglobulin light chain amyloidosis: 2016 update on diagnosis, prognosis, and treatment. Am J Hematol 2016; 91:947–956.
- Patel KS, Hawkins PN. Cardiac amyloidosis: where are we today? J Intern Med 2015; 278:126–144.
- Ritts AJ, Cornell RF, Swiger K, Singh J, Goodman S, Lenihan DJ. Current concepts of cardiac amyloidosis: diagnosis, clinical management, and the need for collaboration. Heart Fail Clin 2017; 13:409–416.
- Sperry BW, Vranian MN, Hachamovitch R, et al. Subtype-specific interactions and prognosis in cardiac amyloidosis. J Am Heart Assoc 2016; 5:e002877.
- Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol 2012; 30:989–995.
- Phelan D, Collier P, Thavendiranathan P, et al. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart 2012; 98:1442–1448.
- Patel AR, Kramer CM. Role of cardiac magnetic resonance in the diagnosis and prognosis of nonischemic cardiomyopathy. JACC Cardiovasc Imaging 2017; 10(10 Pt A):1180–1193.
- White JA, Kim HW, Shah D, et al. CMR imaging with rapid visual T1 assessment predicts mortality in patients suspected of cardiac amyloidosis. JACC Cardiovasc Imaging 2014; 7:143–156.
- Parkey RW, Bonte FJ, Meyer SL, et al. A new method for radionuclide imaging of acute myocardial infarction in humans. Circulation 1974; 50:540–546.
- Wizenberg TA, Muz J, Sohn YH, Samlowski W, Weissler AM. Value of positive myocardial technetium-99m-pyrophosphate scintigraphy in the noninvasive diagnosis of cardiac amyloidosis. Am Heart J 1982; 103(4 Pt 1):468–473.
- Bokhari S, Castaño A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. 99mTc-Pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging 2013; 6:195–201.
- Vranian MN, Sperry BW, Hanna M, et al. Technetium pyrophosphate uptake in transthyretin cardiac amyloidosis: associations with echocardiographic disease severity and outcomes. J Nucl Cardiol 2017; Jan 3. [Epub ahead of print] doi: 10.1007/s12350-016-0768-9.
- Bennett MK, Gilotra NA, Harrington C, et al. Evaluation of the role of endomyocardial biopsy in 851 patients with unexplained heart failure from 2000-2009. Circ Heart Fail 2013; 6:676–684.
- Garcia Y, Collins AB, Stone JR. Abdominal fat pad excisional biopsy for the diagnosis and typing of systemic amyloidosis. Hum Pathol 2017; Nov 10. [Epub ahead of print] doi:10.1016/j.humpath.2017.11.001.
- Quarta CC, Gonzalez-Lopez E, Gilbertson JA, et al. Diagnostic sensitivity of abdominal fat aspiration in cardiac amyloidosis. Eur Heart J 2017; 38:1905–1908.
- Fine NM, Arruda-Olson AM, Dispenzieri A, et al. Yield of noncardiac biopsy for the diagnosis of transthyretin cardiac amyloidosis. Am J Cardiol 2014; 113:1723–1727.
- Katzmann JA, Abraham RS, Dispenzieri A, Lust JA, Kyle RA. Diagnostic performance of quantitative kappa and lambda free light chain assays in clinical practice. Clin Chem 2005; 51:878–781.
- Malha L, Mann SJ. Loop diuretics in the treatment of hypertension. Curr Hypertens Rep 2016; 18:27.
- Rubinow A, Skinner M, Cohen AS. Digoxin sensitivity in amyloid cardiomyopathy. Circulation 1981; 63:1285–1288.
- Feng D, Syed IS, Martinez M, et al. Intracardiac thrombosis and anticoagulation therapy in cardiac amyloidosis. Circulation 2009; 119:2490–2497.
- Castaño A, Drachman BM, Judge D, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev 2015; 20:163–178.
- Hamon D, Algalarrondo V, Gandjbakhch E, et al. Outcome and incidence of appropriate implantable cardioverter-defibrillator therapy in patients with cardiac amyloidosis. Int J Cardiol 2016; 222:562–568.
- Patel KS, Hawkins PN, Whelan CJ, Gillmore JD. Life-saving implantable cardioverter defibrillator therapy in cardiac AL amyloidosis. BMJ Case Rep 2014; Dec 22. doi:10.1136/bcr-2014-206600.
- Gertz M, Lacy M, Dispenzieri A, et al. Troponin T level as an exclusion criterion for stem cell transplantation in light-chain amyloidosis. Leuk Lymphoma 2008; 49:36–41.
- Vranian MN, Sperry BW, Valent J, Hanna M. Emerging advances in the management of cardiac amyloidosis. Curr Cardiol Rep 2015; 17:100.
- Alexander KM, Singh A, Falk RH. Novel pharmacotherapies for cardiac amyloidosis. Pharmacol Ther 2017; 180:129–138.
- Sher T, Fenton B, Akhtar A, Gertz MA. First report of safety and efficacy of daratumumab in 2 cases of advanced immunoglobulin light chain amyloidosis. Blood 2016; 128:1987–1989.
- Sanchorawala V, Palladini G, Kukreti V, et al. A phase 1/2 study of the oral proteasome inhibitor ixazomib in relapsed or refractory AL amyloidosis. Blood 2017; 130:597–605.
- Zumbo G, Sadeghi-Alavijeh O, Hawkins PN, Fontana M. New and developing therapies for AL amyloidosis. Expert Opin Pharmacother 2017; 18:139–149.
- Koyama J, Falk RH. Prognostic significance of strain Doppler imaging in light-chain amyloidosis. JACC Cardiovasc Imaging 2010; 3:333–342.
- Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol 2012; 30:4541–4549.
- Sousa M, Monohan G, Rajagopalan N, Grigorian A. Heart transplantation in cardiac amyloidosis. Heart Fail Rev 2017; 22:317–327.
- Gertz MA, Landau H, Comenzo RL, et al. First-in-human phase I/II study of NEOD001 in patients with light chain amyloidosis and persistent organ dysfunction. J Clin Oncol 2016; 34:1097–1103.
- Holmgren G, Steen L, Ekstedt J, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet 1991; 40:242–246.
- Hanna M. Novel drugs targeting transthyretin amyloidosis. Curr Heart Fail Rep 2014; 11:50–57.
- Berk JL, Suhr OB, Obici L, et al; Diflunisal Trial Consortium. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013; 310:2658–2667.
- Castaño A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail 2012; 18:315–319.
- Sekijima Y, Tojo K, Morita H, Koyama J, Ikeda S-I. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid 2015; 22:79–83.
- Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012; 79:785–792.
- Maurer MS, Grogan DR, Judge DP, et al. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ Heart Fail 2015; 8:519–526.
- Cardoso I, Martins D, Ribeiro T, Merlini G, Saraiva MJ. Synergy of combined doxycycline/TUDCA treatment in lowering transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med 2010; 8:74.
- Obici L, Cortese A, Lozza A, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: A phase II study. Amyloid 2012; 19(suppl 1):34–36.
- Kristen AV, Lehrke S, Buss S, et al. Green tea halts progression of cardiac transthyretin amyloidosis: An observational report. Clin Res Cardiol 2012; 101:805–813.
- Higaki JN, Chakrabartty A, Galant NJ, et al. Novel conformation-specific monoclonal antibodies against amyloidogenic forms of forms of transthyretin. Amyloid 2016; 23:86–97.
WHAT IS AMYLOIDOSIS?
Amyloidosis is a protein deposition disease in which a specific precursor protein pathologically misfolds from its physiologic tertiary structure into a more linear shape dominated by beta-pleated sheets. The misfolded protein aggregates into oligomers, eventually forming insoluble amyloid fibrils that deposit extracellularly in tissues. Both the circulating oligomers, which are cytotoxic, and the fibrils, which cause distortion of the tissue architecture, lead to organ dysfunction. Amyloid fibrils are rigid, nonbranching structures, 7 to 10 nanometers in diameter, with a characteristic appearance on electron microscopy. Affinity for Congo red staining, which binds to the beta-pleated sheets, produces the pathognomonic “apple-green” birefringence when visualized under polarized light microscopy. Universal to all amyloid fibrils are chaperone proteins such as serum amyloid P (SAP) and glycosaminoglycans, as well as calcium. There are more than 30 different precursor proteins implicated in various amyloid diseases, arising as hereditary or nonhereditary, localized or systemic, with different organ involvement and prognosis.1–3
TWO MAIN TYPES OF CARDIAC AMYLOIDOSIS
Light chain amyloidosis (AL)
AL, formerly called primary amyloidosis, is a clonal plasma cell disorder due to the overproduction and misfolding of antibody light chain fragments. It is a rare disease with about 3,000 new cases per year in the United States.5 The median age at diagnosis is 63, although it can present in patients in their 30s and 40s.5,6 It is a systemic disease that often affects the heart, but it can affect several other organs, most commonly the kidneys, gastrointestinal (GI) tract, and nervous system.7
AL is a more aggressive disease than ATTR, with a median untreated survival of less than 6 months in patients who present with heart failure.8 Early diagnosis is crucial as mortality is high without prompt treatment.
Transthyretin amyloidosis (ATTR)
ATTR is due to misfolding of the liver-derived precursor protein transthyretin (TTR) (previously called prealbumin), either as an acquired wild-type variant (ATTRwt) or as a hereditary mutant variant (ATTRm). ATTRwt, known previously as senile CA, typically affects older males and presents as a late onset hypertrophic restrictive cardiomyopathy, often preceded by carpal tunnel syndrome or spinal stenosis or both. The ATTRm variant, caused by one of many different point mutations in the TTR gene, can manifest as a polyneuropathy, cardiomyopathy, or a mixed phenotype that varies according to the specific mutation.
While ATTR portends a better prognosis than AL, it is still a progressive disorder with significantly reduced survival and quality of life. The median survival of patients with the ATTRwt variant is about 4 years and for patients with the ATTRm variant, survival depends on the mutation.9 TTR is a protein tetramer composed of 4 identical 127-amino acid monomers noncovalently bound at a dimer-dimer interface (Figure 1). It is a transport protein for thyroxine and retinol binding protein. The dissociation of the tetramer is the rate-limiting step for amyloid fibrillogenesis. Differentiating the ATTRwt variant and the ATTRm variant is done by testing the TTR gene for a mutation.1,3
How common is the ATTRwt variant? The ATTRwt variant is often an unrecognized cause of diastolic heart failure in the elderly, with up to 25% of patients 85 and older showing ATTRwt amyloid deposits on autopsy studies.10 A recent study showed that 13% of patients 60 and older hospitalized with heart failure with preserved ejection fraction had grade 2 to 3 uptake on 99mtechnetium-pyrophosphate (99mTcPYP) scintigraphy, which is consistent with ATTR-CA.11 In 43 consecutive patients undergoing transcatheter aortic valve replacement, 11.6% were found to have significant uptake on 99mTcPYP scan.12 It is clear given the aging population that the ATTRwt variant will become the most common form of amyloidosis. It is much more common in white males, with a median age at diagnosis of 75.13 Carpal tunnel syndrome (almost always bilateral) and spinal stenosis are present in about 50% of patients diagnosed with ATTRwt-CA and often precede clinical presentation of heart failure by 5 to 15 years.14–17
How common is the ATTRm variant? There are more than 100 point mutations in the TTR gene that lead to various familial TTR-related amyloid syndromes, either neuropathic (familial amyloid polyneuropathy [FAP]) or cardiomyopathic (familial amyloid cardiomyopathy).18 The most common mutation in the United States is V122I in which there is an isoleucine substitution for valine at the 122nd amino acid position. This mutation is seen in African Americans, 3% to 4% of whom are heterozygote carriers.19 Although the true penetrance is unknown, this mutation can lead to a late-onset restrictive cardiomyopathy with minimal neuropathy and is frequently misdiagnosed as hypertensive heart disease or diastolic heart failure. The median survival for V122I ATTRm-CA is about 2 years but likely depends on the stage at the time of diagnosis.20 The second most common mutation in the United States, T60A, is seen in patients of Irish descent and causes a mixed neuropathy and cardiomyopathy.17
PATHOLOGY AND PATHOPHYSIOLOGY OF CA
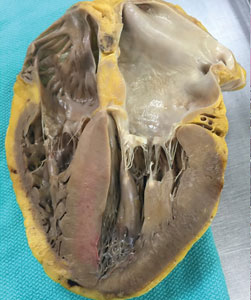
The atria are universally involved with interatrial septal thickening, which can lead to poor atrial function and increased rates of atrial fibrillation (ATTR more so than AL).21,29 The conduction system can be affected causing varying degrees of heart block, as well as bundle branch block (ATTR more so than AL).30 The valves are usually thickened, often associated with mild to moderate regurgitation. Pericardial involvement can lead to small pericardial effusions (large effusions are rare), and coronary involvement (classically, small intramural vessels) can lead to ischemia and angina with normal epicardial coronaries (AL more so than ATTR).31–33
Thickened left and right ventricular walls result in a nondilated ventricle that is stiff and poorly compliant, resulting in progressive diastolic filling abnormalities. Systolic dysfunction can be seen in severe and advanced disease. Importantly, ejection fraction measured by echocardiography is misleading in CA, as reduced end-diastolic volume produces a low stroke volume. For example, an ejection fraction of 50%, when starting at a significantly reduced end-diastolic volume (for example, 70 mL), leads to a significantly reduced stroke volume (35 mL) and, thus, cardiac output. This explains why patients with CA cannot usually tolerate reduced heart rates, as their cardiac output is dependent on heart rate.34–36
CLINICAL PRESENTATION
Patients with CA typically exhibit heart failure with preserved ejection fraction (otherwise known as diastolic heart failure). Dyspnea on exertion is common; however, some patients can present with more right-sided heart failure symptoms such as lower-extremity edema and ascites. Fatigue and weakness are related to low cardiac output and often attributed to nonspecific symptoms of aging. Because of the thickened ventricles, patients can often be misdiagnosed as having HCM with or without obstruction.7,34 The first manifestation of CA may be atrial fibrillation, most commonly in ATTRwt-CA, or cardioembolic stroke. Atrial fibrillation can be present for years before CA is considered. Bundle branch block and complete heart block (more common in ATTR-CA than AL-CA) may lead to pacemaker implantation.30 Angina with normal coronaries can occur, and a rare presentation may be cardiogenic shock due to diffuse ischemia.31–33 Elderly patients with CA can present with low-flow, low-gradient aortic stenosis.37
DIAGNOSIS
ECG
As opposed to that seen in true left ventricular hypertrophy (LVH), which leads to increased voltage on ECG, amyloid infiltration of the myocardium leads to lower voltage. Thus, what is indicative of LVH on echocardiogram combined with low voltage on the ECG is a classic finding for CA. However, only about 50% of patients with AL-CA and about 30% of patients with ATTR-CA meet true low-voltage criteria (QRS amplitude less than 5 mm in limb leads or less than 10 mm in precordial leads).30,38 Hence, the absence of low-voltage criteria does not exclude the diagnosis of CA. Approximately 10% of patients with CA confirmed by biopsy met ECG criteria for LVH.38 The key point is to consider the overall degree of voltage on the ECG relative to the degree of LV thickening on the echocardiogram, recognizing that lower voltage than what would be expected may indicate possible infiltrative disease such as CA. The other main finding on the ECG in patients with CA is a pseudoinfarct pattern with Q waves in the early precordial leads mimicking a prior anteroseptal myocardial infarction.38,39 This finding is seen in about 50% of patients (Figure 3).39 Wide QRS complexes are more frequent in ATTR-CA and lower limb voltages are more frequent in AL-CA.30
Echocardiogram
The echocardiographic finding of LVH in patients with CA is misleading in that the LV thickening is due to infiltrating amyloid fibrils and not to myocyte hypertrophy. That said, the terms LVH and LV thickening are used interchangeably when describing the echocardiographic phenotype. LV wall thickness greater than 12 mm (6 mm to 10 mm is normal) in the absence of hypertension should prompt suspicion for CA.34 LV thickening most often appears symmetric; however, occasionally it may exhibit asymmetric septal hypertrophy, particularly in ATTRwt-CA. In some cases, there may be a smaller subset that can actually have dynamic LV outflow obstruction similar to that seen in hypertrophic obstructive cardiomyopathy.22–24 An important echocardiographic clue that can differentiate CA from other diseases is thickening of both the LV and right ventricle (Figure 3). Septal wall thickness and LV mass index are greater in ATTR-CA compared with AL-CA.30 On average, the LV septum is around 15 mm in AL-CA and around 18 mm in ATTRwt-CA.40 Historically, the characteristic myocardial “granular sparking” or “speckling” pattern has low sensitivity and specificity.16 The left ventricle is not dilated; rather, the ventricular dimensions are usually smaller than normal. Although ejection fraction is usually preserved, cardiac output is low due to decreased ventricular volume.37 Systolic dysfunction occurs late in the disease.16 Diastolic dysfunction is universal, with a mitral inflow pattern that can range from stage I (abnormal relaxation) in early disease to stage III (restrictive filling pattern) in more advanced disease. Septal and lateral tissue Doppler velocities are very low in amyloid heart disease.41 Another echocardiographic clue to diagnosis is thickening of the heart valves, which is not seen in hypertensive heart disease or hypertrophic cardiomyopathy. Biatrial dilation is common, and there can be thickening of the interatrial septum.
Laboratory testing
N-terminal pro-b-type natriuretic peptide (NT-proBNP) is universally elevated in CA and is typically higher in AL-CA than in ATTR-CA. Troponin T or troponin I or both may be chronically elevated in CA and likely signify small-vessel ischemia. In the appropriate clinical context of a thickened ventricle and heart failure, an elevated troponin value (outside of an acute coronary syndrome) should trigger suspicion for CA. Workup for a monoclonal protein process should always be done when considering CA to rule out AL. Serum and urine protein electrophoresis are insensitive tests to detect AL and should not be relied upon as a screening test. The serum free light chain (sFLC) assay, which measures free kappa and lambda light chain levels and reports the ratio, is a sensitive test that should be measured routinely along with immunofixation of the serum and urine. In AL, sFLC will reveal an abnormal kappa-lambda ratio. An abnormally low ratio (less than 0.26) suggests a monoclonal lambda light chain process, while an abnormally high ratio (greater than 1.65) suggests a monoclonal kappa light chain process. Immunofixation will reveal an M-protein. Because light chains are excreted by the kidney, the serum levels of both kappa and lambda will be elevated in renal dysfunction, but the ratio should remain normal.16,31,34,40,42–47
Advanced noninvasive diagnostic tools for CA
Over the past decade, the ability to diagnose CA noninvasively has dramatically improved with strain imaging using 2D speckle tracking echocardiography, cardiac magnetic resonance imaging (MRI), and nuclear bone scintigraphy. These diagnostic tools have given clinicians options to pursue the diagnosis of CA without directly proceeding to endomyocardial biopsy.
Cardiac MRI. Cardiac MRI is useful for the diagnosis of CA (Figure 4B, 4C). Imaging after administration of gadolinium contrast shows a characteristic late gadolinium enhancement (LGE) pattern that is diffuse and subendocardial, and does not follow any particular coronary distribution.49 LGE can also be seen in the right ventricle and the atrial walls, and can be transmural and patchy in ATTRwt-CA. This pattern is highly sensitive (93%) and specific (70%) for CA with an overall negative predictive accuracy of 84%.50
One of the main limitations of cardiac MRI for the diagnosis of CA is the inability to give contrast in patients with reduced glomerular filtration rate. However, native T1-myocardial mapping techniques that do not require contrast show significantly increased native T1 times in CA and offer a promising alternative. Cardiac MRI parameters such as LGE, the difference in inversion time between the LV cavity and myocardium, native T1 mapping, and extracellular volume offer prognostic information. A greater than fivefold mortality increase is seen in CA patients with transmural LGE compared with those without LGE.49,50
99mTcPYP scintigraphy. 99mTcPYP, a radiotracer used in bone scans, was initially used in cardiology to quantify myocardial infarction due to its ability to localize calcium.51 Its potential utility in CA came in 1982 when diffuse myocardial 99mTcPYP uptake on cardiac radionucleotide imaging was noted in 10 patients with tissue-proven amyloidosis.52 Several subsequent studies reproduced and expanded upon this observation and revealed its diagnostic value, specifically showing that there is significant uptake in ATTR-CA and no to mild uptake in AL-CA. This offers a significant advantage over other noninvasive modalities in that it not only confirms the diagnosis of CA but differentiates ATTR-CA.16
99mTcPYP myocardial radiotracer uptake is graded by the semiquantitative visual score of cardiac retention, where grade 0 = no cardiac uptake, grade 1 = mild uptake less than bone, grade 2 = moderate uptake equal to bone, and grade 3 = high uptake greater than bone (Figure 4D).42 Additionally, quantitative analysis of heart retention can be calculated drawing circular regions of interest over the heart and mirrored on the contralateral chest wall. A heart-to-contralateral ratio greater than 1.5 is consistent with the diagnosis of ATTR-CA.53,54 In 2016, a multicenter study showed that grade 2 or 3 myocardial radiotracer uptake on bone scintigraphy in the absence of evidence of a monoclonal gammopathy was diagnostic for ATTR-CA, providing a cost-effective and non-invasive technique with a specificity and positive predictive value of 100% (confidence interval, 99.0–100%).42
Endomyocardial biopsy, right heart catheterization, and fat biopsy
Endomyocardial biopsy is essentially 100% sensitive for the diagnosis of CA.25 The main risk of pursuing endomyocardial biopsy is about a 1% risk of right ventricular perforation leading to cardiac tamponade.55 The other limitation to this approach is that not all centers are equipped to perform this procedure. Birefringence under polarized light microscopy is histopathologically diagnostic of CA; however, further subtyping by the pathologist to determine if it is AL or ATTR is absolutely crucial. Subtyping can be performed by immunohistochemistry with caution taken for misinterpretation. If there is any question of accuracy, the specimen should be sent for laser microdissection and mass spectroscopy for accurate identification of the precursor protein type (some centers routinely perform mass spectroscopy on all myocardial specimens).16,31
Right heart catheterization is nonspecific and shows restrictive hemodynamics. The right atrial waveform shows rapid x and y descents and the right ventriclular tracing may show a dip-and-plateau pattern typical of restrictive cardiomyopathy. Cardiac output can be preserved but more commonly is low.16,31
Fat pad biopsy is 60% to 80% sensitive in AL, 65% to 85% sensitive in ATTRm, and only 14% sensitive in ATTRwt, with the accuracy dependent on the operator, pathologist, and how much tissue is removed (fat pad aspirate vs biopsy specimen).56–58 Fat pad biopsy has diagnostic limitations, and a negative fat pad biopsy does not rule out amyloidosis.
DIAGNOSTIC ALGORITHM FOR CARDIAC AMYLOIDOSIS
In conjunction with laboratory tests, 99mTcPYP scan of the heart can be ordered to investigate the possibility of ATTR-CA. Grade 2 to 3 myocardial uptake in the absence of a monoclonal plasma cell process is consistent with the diagnosis of ATTR-CA. Grade 0 or 1 myocardial uptake on 99mTcPYP scan with an abnormal sFLC ratio or positive M protein on immunofixation suggests AL-CA and a bone marrow biopsy should be performed. If the patient has an abnormal sFLC ratio and grade 2 to 3 uptake on 99mTcPYP scan, the diagnosis of ATTR-CA with unrelated monoclonal gammopathy of undetermined significance should be considered. However, this would need to be reconciled by pursuing endomyocardial biopsy and accurate tissue typing. If the 99mTcPYP scan is negative, and the sFLC ratio is normal, and immunofixation is negative, a diagnosis of CA is very unlikely.16,31,40,42–47
If the diagnosis of ATTR-CA is made, genetic testing can determine the presence or absence of a mutation to differentiate ATTRm or ATTRwt, respectively. If the diagnosis of AL-CA is suggested, a bone marrow biopsy is necessary to identify and quantify the plasma cell clone.16,31,40,42–47
TREATMENT
Management of heart failure in cardiac amyloidosis
The main treatment of heart failure revolves around sodium restriction and diuretics to relieve congestion. This can prove challenging in many patients due to the narrow window between too high or too low filling pressures. A combination of loop diuretics and an aldosterone antagonist is most effective.31,34,44,45 Torsemide is preferred over furosemide due to its superior bioavailability and longer duration of action, particularly since these patients have issues with gut edema and GI absorption. Due to dependence of the cardiac output on heart rate and the tendency for orthostatic hypotension, traditional neurohormonal antagonists including beta blockers and angiotensin-converting enzyme inhibitors are neither effective nor well tolerated.60 However, in patients with atrial fibrillation, beta blockers may need to be used for rate control. Nondihydropyridine calcium channel blockers bind avidly to amyloid fibrils and are contraindicated due to risk of profound hypotension and syncope.30,31,34,44,45 Digoxin is usually avoided in CA due to concerns of increased risk of toxicity; however, it may be used with caution for rate control in atrial fibrillation given its lack of negative inotropy.61 Maintenance of normal sinus rhythm is preferable due to the importance of atrial contribution to cardiac output.
Anticoagulation in patients with atrial fibrillation and even in patients with normal sinus rhythm and poor atrial function is important due to the high risk of thromboembolic complications.62 Pacemakers are indicated for heart block or symptomatic bradycardia.63 The role of intracardiac defibrillators is controversial, but may be warranted in selected patients with AL-CA.64,65
AL treatment
Risk stratification and prognostication for AL. The most important determinant of clinical outcome in AL is the extent of cardiac involvement, as congestive heart failure and sudden cardiac death are the most common causes of death. The level of NT-proBNP and the level of either troponin T or troponin I have strong prognostic value and form the basis for the staging system in AL. Various iterations have evolved over the years, but the most widely adopted is the 4-stage system developed and validated by Mayo Clinic. This system uses a cutoff value at diagnosis for NT-proBNP greater than 1,800 ng/mL, troponin T greater than 0.025 μg/L, and the difference between kappa and lambda free light chain levels greater than 180 mg/L. Stage level increases by the number of cutoff values exceeded, with stage IV carrying a median survival of 6 months.47 Additionally, the troponin T level can help risk-stratify patients being considered for autologous stem cell transplant. In a retrospective study, troponin T greater than 0.06 μg/L was associated with increased mortality following stem cell transplant.66 Ultimately, prognosis in AL-CA is related to the hematologic response to chemotherapy.
Current treatment strategies for AL. The survival of patients with AL has improved over the years with the advent of more effective chemotherapeutic regimens that kill the underlying plasma cell clone producing the unstable light chains. The goal of treatment is to achieve a complete hematologic response with normalization of the affected light chain and sFLC ratio as well as elimination of the M protein on immunofixation.
The development of the proteasome inhibitor bortezomib has improved efficacy and survival in AL causing a faster and more complete hematologic response than prior regimens.43,67,68
The most commonly used first-line treatment consists of a 3-drug combination with the alkylating agent cyclophosphamide, the proteasome inhibitor bortezomib, and the steroid dexamethasone, which is given weekly.43 A retrospective study by Sperry et al8 showed that patients receiving an alkylating agent, bortezomib, and a steroid had the best outcomes compared with other regimens.
For patients with refractory or relapsed disease, the CD38 monoclonal antibody daratumumab can be used if patients meet myeloma criteria and has been found to be effective thus far.68,69 Newer proteasome inhibitors such as ixazomib, which is taken orally, are being studied in alternative combination regimens.70 High-dose chemotherapy with autologous stem cell transplant can be considered in patients with an acceptable cardiac risk profile and may offer more complete and durable remission than chemotherapy, although this is controversial.43,71
The cardiologist’s role in AL-CA. The hematologist directs the chemotherapy for AL but works closely with the cardiologist when there is cardiac involvement. The main role of the cardiologist is to manage volume status with diuretics, monitor for arrhythmia, and evaluate the cardiac response to treatment.43,71 Cardiac response was traditionally measured by echocardiographic changes of wall thickness, diastolic function, and ejection fraction, as well as changes in New York Heart Association (NYHA) functional class.67 However, it is uncommon to see reduction in LV wall thickness or significant improvement in ejection fraction and if it does occur, it is a slow process that usually takes more than 1 year. With the advent of longitudinal myocardial strain imaging, improvements in strain can be seen despite the lack of structural changes on echocardiogram.72 In 2012, consensus criteria defined a cardiac response as a greater than 30% reduction in NT-proBNP.73 Misfolded light chains are toxic to cardiomyocytes by causing increased oxidative stress and impairing contractility. Thus, reduction in light chain levels can lead to clinical improvement and significant reductions in NT-proBNP without changing amyloid fibril burden in the heart.
Heart transplant for patients with AL-CA. For patients who have a good hematologic response to initial chemotherapy but have limited predicted survival due to severe heart failure, heart transplant followed by autologous stem cell transplant, is a treatment strategy that can be considered. The patient must have clinically isolated severe cardiac disease, minimal amyloid burden in other organs, and a plasma cell clone that is responsive to therapy. Initial reports of heart transplant showed poor survival rates due to recurrent amyloid in the transplanted heart and progressive amyloid deposition in other organs. However, due to improved anti-plasma-cell-directed therapy and refinement in patient selection, outcomes have improved. Contemporary series of patients undergoing heart transplant followed by stem cell transplant showed that outcomes are almost comparable to heart transplant for other indications, with a 5-year survival rate of approximately 65%.74
Future therapies for AL. There is an AL amyloid-directed monoclonal antibody designed to remove amyloid fibrils from affected organs and is currently undergoing clinical trials. NEOD001, a humanized murine monoclonal antibody that targets an epitope exposed during light chain misfolding, binds to the light chain amyloid fibril and signals an immune response to clear the deposits. This agent has completed phase 1 and 2 clinical trials of 27 patients previously treated with at least 1 plasma cell-directed therapy. It showed good tolerability and achieved both renal and cardiac responses in most patients.75
A phase 2b clinical trial (NCT02632786) of patients with AL with a previous hematologic response to treatment and persistent heart dysfunction is underway and expected to be completed in January 2018. A phase 3 clinical trial (NCT02312206) of NEOD001 as an adjunct to chemotherapy is also ongoing and results are expected in February 2019.
ATTR treatment
Liver transplant for ATTRm amyloidosis for the V30M mutation that causes FAP was first described in 1990, but it has not been well validated in other mutations and is not a solution for ATTRwt.76 Significant progress has been made over the past 2 decades in the understanding of the pathophysiologyy of ATTR, paving the way for promising advancements in pharmacotherapy. Presently, there are 3 classes of pharmacologic agents, grouped by the point of disease process each strategy targets:
- Block TTR synthesis at the translational level in hepatocytes
- Stabilize the TTR tetramer to inhibit the rate-determining step of amyloidogenesis
- Disrupt and clear the ATTR amyloid fibril.77
Block TTR synthesis. TTR messenger RNA (mRNA) can be targeted by “silencers” preventing translation, thereby reducing the production TTR protein by hepatocytes. The resultant sustained reduction of plasma TTR should decrease or halt amyloid deposition by making less TTR available to dissociate and deposit in the heart and nerves. There are 2 approaches to silencing TTR mRNA translation: small interfering RNA (siRNA) and antisense oligonucleotide (ASO).77
An siRNA, packaged in a lipid nanoparticle to ensure delivery to the liver, has been designed to bind to a conserved region of TTR mRNA, degrading the mRNA and reducing TTR protein expression. One siRNA, patisiran, completed the phase 3 APOLLO clinical trial (NCT01960348) in August 2017. It is an intravenous medication that requires premedication. The APOLLO trial studied patients with neuropathic variants of ATTRm, with a primary end point of neuropathy progression over 18 months. Patisrian met the primary end point and will likely be approved by the US Food and Drug Administration (FDA) by the third quarter of 2018. Given its target of a conserved 3’ untranslated region of TTR mRNA, patisiran should theoretically yield benefits not just in FAP but to both ATTRm-CA and ATTRwt-CA.68,77
ASOs are single-stranded oligonucleotides, typically 20 nucleotides in length, that bind to mRNA and elicit enzymatic mRNA degradation and reduced protein expression. Inotersen (IONIS-TTRRx) is an ASO drug that targets a conserved region of TTR mRNA. Inotersen is administered by weekly subcutaneous injection. A phase 2/3 clinical trial (NCT01737398) completed in October 2017 studied the drug's efficacy in treating FAP, with a primary end point of neuropathy progression over 65 weeks. It met the primary end point and a subset of patients actually improved. Like patisiran, inotersen is likely to be approved by the FDA for a neuropathy indication by the third quarter of 2018 and may benefit patients with ATTRm-CA and ATTRwt-CA. More studies are needed in the ATTR cardiac population.68,77
Stabilize the TTR tetramer. The TTR tetramer has 2 thyroxine binding pockets that stabilize the structure when bound preventing dissociation. Dissociation of the tetramer, the rate limiting step for ATTR fibrillogenesis, can be reduced using pharmacologic agents that bind to the thyroxine binding pockets. Both new and repurposed agents have been found to stabilize the TTR tetramer, including diflunisal, tafamidis, tolcapone, and AG10.68,77
Diflunisal (Dolobid) is a nonacetylated salicylate nonsteroidal anti-inflammatory drug used for over 3 decades to treat arthritis and musculoskeletal pain. Unrelated to its anti-inflammatory properties, it interacts with TTR’s thyroxine binding pocket to increase the stability of the tetramer. A 2013 randomized, placebo-controlled, international multicenter trial of 130 patients with FAP demonstrated a statistically significant slowing of polyneuropathy progression; however, 67 patients (diflunisal n = 27, placebo n = 40) did not complete the study.78 A small study revealed diflunisal to be reasonably well-tolerated in ATTR-CA, and to date it is the only readily available pharmacotherapy for ATTR supported by a randomized, placebo-controlled trial.79,80 Diflunisal may be considered for off-label use in patients with ATTR-CA with relatively preserved kidney function and no increased bleeding risk, taken under the supervision of a cardiologist who will monitor for fluid retention and changes in renal function.77
Tafamidis (Vyndaqel), like diflunisal, interacts with TTR’s thyroxine binding pocket and increases tetrameric stability. A phase 2 open-label clinical trial studied the efficacy and tolerance of tafamidis in 31 patients with ATTRwt and NYHA functional class 1 and 2 followed for 1 year. Twenty-eight patients completed the study and 2 patients died. TTR stabilization at 6 weeks was achieved in 30 of 31 patients (96.8%), and success at 1 year in 25 of 28 patients (89.3%). No clinical progression occurred in 16 of 31 patients (51.5%), and tafamidis was generally well-tolerated, with diarrhea the most common side effect in 7 of 31 patients (22.6% ).81,82
A phase 3 clinical trial (NCT01994889) is studying tafamidis vs placebo in patients with ATTRm-CA or ATTRwt-CA (excluding NYHA functional class 4) with the primary outcome measure of all-cause mortality and heart failure-related hospitalizations over 30 months. This is a large trial that enrolled 446 patients and data collection is expected to be completed in February 2018.
Tolcapone, approved by the FDA for Parkinson disease, has been found to be a potent TTR stabilizer by binding both thyroxine binding pockets of the TTR tetramer simultaneously. However, tolcapone has an FDA “black box” warning due to the risk of potentially fatal acute fulminant liver failure and is not currently used in ATTR therapy.68 Nonetheless, it will likely undergo further study for ATTR.
Recruiting is underway for a phase 1 clinical trial of AG10, a potent selective TTR stabilizer (NCT03294707).68
Disruption and clearance of the ATTR amyloid fibril. Even though treatment directed at blocking TTR synthesis or stabilizing the tetramer may be effective at preventing further deposition, the residual amyloid deposits persist and continue to affect organ function. With that need in mind, several agents that disrupt the amyloid formation process further downstream have been evaluated at a basic science level and in a few small nonrandomized open-label studies.77
Doxycycline is a tetracycline antibiotic with demonstrated effectiveness in disrupting mature amyloid fibrils in mouse models.83 Tauroursodeoxycholic acid is a bile acid with the ability to disrupt prefibrillar amyloid components. This combination has been studied in patients with ATTR-CA in 2 small open-label trials, with only 1 having results published. There was no progression in NT-proBNP or wall thickness in a cohort of 7 patients who completed 12 months of treatment.84 These data are nonrandomized and hypothesis-generating, as adequate studies would need to be performed to evaluate this hypothesis further. Because there are currently no FDA-approved therapies for ATTR, patients may be offered this combination fully informed that the data are limited. (Note: ursodiol is substituted for tauroursodeoxycholic acid, which is not available in the United States.)68
Green tea extract contains the polyphenol epigallocatechin-3-gallate, which has shown the ability for fibril disruption as well as TTR stabilization, importantly using a binding site separate from the thyroxine binding pocket (utilized in diflunisal and tafamidis). A small open-label study of 19 patients with ATTR-CA of whom 14 took green tea extract for 1 year reported a reduction in intraventricular septal wall thickness at 1 year, and in 9 patients a reduction of 12.5% in LV mass measured by cardiac MRI.85
Curcumin, the active ingredient in the household spice turmeric, has displayed in vitro promise as a TTR stabilizer by binding to the thyroxine binding pocket, and as an amyloid fibril disruptor by increasing macrophage degradation activity. Although there have only been preliminary animal studies, this supplement may be promising for further study in humans with ATTR-CA.68
PRX004 is a synthetic antibody designed to bind to non-native misfolded forms of TTR with the goal of potentially preventing deposition and promoting clearance of TTR aggregates.86 A phase 1 open-label escalation trial (NCT03336580) in 36 patients with ATTRm is planned and, hopefully, will pave the way for further study.
Heart transplant for patients with ATTR-CA. Patients with ATTRwt-CA who are young enough to undergo heart transplant have displayed favorable outcomes given that it causes clinically isolated heart disease and is an indolent process that should not affect the transplanted heart over the average life span of the allograft. Patients with the ATTRm mutation V122I have been treated with heart transplant alone, with the thought process that again, due to the indolent nature of amyloid deposition, concomitant liver transplant may not be needed.74 Thus far, 6 patients with this mutation have undergone successful transplant at our institution with heart alone, 1 of whom is 9 years posttransplant without any recurrent amyloid in the allograft. Patients with the T60A mutation that causes both polyneuropathy and cardiomyopathy require combined heart and liver transplant.74
Are there other therapies being studied to clear amyloid deposits and reverse organ dysfunction?
Extracellular deposits of amyloid fibrils, regardless of precursor protein, contain common elements such as calcium, glycosaminoglycans, and an SAP component. SAP stabilizes amyloid fibrils and makes them resistant to degradation. A monoclonal immunoglobulin G1 anti-SAP antibody has been designed to target the ubiquitous SAP component, signaling an immune response that leads to macrophage mediated clearance of amyloid fibrils, regardless of the type. Treatment with this approach was studied in a pilot trial of 16 patients mostly with AL, and within a 6-week period some of the patients had dramatic reversal of liver amyloid deposition.2 This has paved the way for a phase 2 open-label trial to be performed in patients with AL-CA and ATTR-CA (NCT03044353) and plans for a randomized phase 3 trial in CA are in discussion. There is optimism that this therapy may achieve the holy grail of removing amyloid rather than just preventing further deposition.
CONCLUSION
The diagnosis of CA requires a high index of suspicion. The diagnostic tools have improved due to the availability of modern imaging techniques, and the advent of measuring the sFLC assay along with immunofixation of the serum and urine. The prognosis for patients with AL-CA used to be dismal, with very poor survival rates. The current treatment strategies that include proteasome inhibitors have significantly improved survival, emphasizing the importance of early diagnosis and prompt initiation of therapy. Monoclonal antibodies against plasma cells (daratumumab) and light chain amyloid deposits (NEOD001) have the potential to further improve outcomes. The diagnosis of ATTR-CA used to be a futile academic pursuit given the lack of available therapies. However, there are several new FDA-approved agents on the horizon, including TTR gene silencers and stabilizers. CA is no longer considered to be rare and hopeless. Rather, it is more common than previously recognized and even more treatable.
WHAT IS AMYLOIDOSIS?
Amyloidosis is a protein deposition disease in which a specific precursor protein pathologically misfolds from its physiologic tertiary structure into a more linear shape dominated by beta-pleated sheets. The misfolded protein aggregates into oligomers, eventually forming insoluble amyloid fibrils that deposit extracellularly in tissues. Both the circulating oligomers, which are cytotoxic, and the fibrils, which cause distortion of the tissue architecture, lead to organ dysfunction. Amyloid fibrils are rigid, nonbranching structures, 7 to 10 nanometers in diameter, with a characteristic appearance on electron microscopy. Affinity for Congo red staining, which binds to the beta-pleated sheets, produces the pathognomonic “apple-green” birefringence when visualized under polarized light microscopy. Universal to all amyloid fibrils are chaperone proteins such as serum amyloid P (SAP) and glycosaminoglycans, as well as calcium. There are more than 30 different precursor proteins implicated in various amyloid diseases, arising as hereditary or nonhereditary, localized or systemic, with different organ involvement and prognosis.1–3
TWO MAIN TYPES OF CARDIAC AMYLOIDOSIS
Light chain amyloidosis (AL)
AL, formerly called primary amyloidosis, is a clonal plasma cell disorder due to the overproduction and misfolding of antibody light chain fragments. It is a rare disease with about 3,000 new cases per year in the United States.5 The median age at diagnosis is 63, although it can present in patients in their 30s and 40s.5,6 It is a systemic disease that often affects the heart, but it can affect several other organs, most commonly the kidneys, gastrointestinal (GI) tract, and nervous system.7
AL is a more aggressive disease than ATTR, with a median untreated survival of less than 6 months in patients who present with heart failure.8 Early diagnosis is crucial as mortality is high without prompt treatment.
Transthyretin amyloidosis (ATTR)
ATTR is due to misfolding of the liver-derived precursor protein transthyretin (TTR) (previously called prealbumin), either as an acquired wild-type variant (ATTRwt) or as a hereditary mutant variant (ATTRm). ATTRwt, known previously as senile CA, typically affects older males and presents as a late onset hypertrophic restrictive cardiomyopathy, often preceded by carpal tunnel syndrome or spinal stenosis or both. The ATTRm variant, caused by one of many different point mutations in the TTR gene, can manifest as a polyneuropathy, cardiomyopathy, or a mixed phenotype that varies according to the specific mutation.
While ATTR portends a better prognosis than AL, it is still a progressive disorder with significantly reduced survival and quality of life. The median survival of patients with the ATTRwt variant is about 4 years and for patients with the ATTRm variant, survival depends on the mutation.9 TTR is a protein tetramer composed of 4 identical 127-amino acid monomers noncovalently bound at a dimer-dimer interface (Figure 1). It is a transport protein for thyroxine and retinol binding protein. The dissociation of the tetramer is the rate-limiting step for amyloid fibrillogenesis. Differentiating the ATTRwt variant and the ATTRm variant is done by testing the TTR gene for a mutation.1,3
How common is the ATTRwt variant? The ATTRwt variant is often an unrecognized cause of diastolic heart failure in the elderly, with up to 25% of patients 85 and older showing ATTRwt amyloid deposits on autopsy studies.10 A recent study showed that 13% of patients 60 and older hospitalized with heart failure with preserved ejection fraction had grade 2 to 3 uptake on 99mtechnetium-pyrophosphate (99mTcPYP) scintigraphy, which is consistent with ATTR-CA.11 In 43 consecutive patients undergoing transcatheter aortic valve replacement, 11.6% were found to have significant uptake on 99mTcPYP scan.12 It is clear given the aging population that the ATTRwt variant will become the most common form of amyloidosis. It is much more common in white males, with a median age at diagnosis of 75.13 Carpal tunnel syndrome (almost always bilateral) and spinal stenosis are present in about 50% of patients diagnosed with ATTRwt-CA and often precede clinical presentation of heart failure by 5 to 15 years.14–17
How common is the ATTRm variant? There are more than 100 point mutations in the TTR gene that lead to various familial TTR-related amyloid syndromes, either neuropathic (familial amyloid polyneuropathy [FAP]) or cardiomyopathic (familial amyloid cardiomyopathy).18 The most common mutation in the United States is V122I in which there is an isoleucine substitution for valine at the 122nd amino acid position. This mutation is seen in African Americans, 3% to 4% of whom are heterozygote carriers.19 Although the true penetrance is unknown, this mutation can lead to a late-onset restrictive cardiomyopathy with minimal neuropathy and is frequently misdiagnosed as hypertensive heart disease or diastolic heart failure. The median survival for V122I ATTRm-CA is about 2 years but likely depends on the stage at the time of diagnosis.20 The second most common mutation in the United States, T60A, is seen in patients of Irish descent and causes a mixed neuropathy and cardiomyopathy.17
PATHOLOGY AND PATHOPHYSIOLOGY OF CA
The atria are universally involved with interatrial septal thickening, which can lead to poor atrial function and increased rates of atrial fibrillation (ATTR more so than AL).21,29 The conduction system can be affected causing varying degrees of heart block, as well as bundle branch block (ATTR more so than AL).30 The valves are usually thickened, often associated with mild to moderate regurgitation. Pericardial involvement can lead to small pericardial effusions (large effusions are rare), and coronary involvement (classically, small intramural vessels) can lead to ischemia and angina with normal epicardial coronaries (AL more so than ATTR).31–33
Thickened left and right ventricular walls result in a nondilated ventricle that is stiff and poorly compliant, resulting in progressive diastolic filling abnormalities. Systolic dysfunction can be seen in severe and advanced disease. Importantly, ejection fraction measured by echocardiography is misleading in CA, as reduced end-diastolic volume produces a low stroke volume. For example, an ejection fraction of 50%, when starting at a significantly reduced end-diastolic volume (for example, 70 mL), leads to a significantly reduced stroke volume (35 mL) and, thus, cardiac output. This explains why patients with CA cannot usually tolerate reduced heart rates, as their cardiac output is dependent on heart rate.34–36
CLINICAL PRESENTATION
Patients with CA typically exhibit heart failure with preserved ejection fraction (otherwise known as diastolic heart failure). Dyspnea on exertion is common; however, some patients can present with more right-sided heart failure symptoms such as lower-extremity edema and ascites. Fatigue and weakness are related to low cardiac output and often attributed to nonspecific symptoms of aging. Because of the thickened ventricles, patients can often be misdiagnosed as having HCM with or without obstruction.7,34 The first manifestation of CA may be atrial fibrillation, most commonly in ATTRwt-CA, or cardioembolic stroke. Atrial fibrillation can be present for years before CA is considered. Bundle branch block and complete heart block (more common in ATTR-CA than AL-CA) may lead to pacemaker implantation.30 Angina with normal coronaries can occur, and a rare presentation may be cardiogenic shock due to diffuse ischemia.31–33 Elderly patients with CA can present with low-flow, low-gradient aortic stenosis.37
DIAGNOSIS
ECG
As opposed to that seen in true left ventricular hypertrophy (LVH), which leads to increased voltage on ECG, amyloid infiltration of the myocardium leads to lower voltage. Thus, what is indicative of LVH on echocardiogram combined with low voltage on the ECG is a classic finding for CA. However, only about 50% of patients with AL-CA and about 30% of patients with ATTR-CA meet true low-voltage criteria (QRS amplitude less than 5 mm in limb leads or less than 10 mm in precordial leads).30,38 Hence, the absence of low-voltage criteria does not exclude the diagnosis of CA. Approximately 10% of patients with CA confirmed by biopsy met ECG criteria for LVH.38 The key point is to consider the overall degree of voltage on the ECG relative to the degree of LV thickening on the echocardiogram, recognizing that lower voltage than what would be expected may indicate possible infiltrative disease such as CA. The other main finding on the ECG in patients with CA is a pseudoinfarct pattern with Q waves in the early precordial leads mimicking a prior anteroseptal myocardial infarction.38,39 This finding is seen in about 50% of patients (Figure 3).39 Wide QRS complexes are more frequent in ATTR-CA and lower limb voltages are more frequent in AL-CA.30
Echocardiogram
The echocardiographic finding of LVH in patients with CA is misleading in that the LV thickening is due to infiltrating amyloid fibrils and not to myocyte hypertrophy. That said, the terms LVH and LV thickening are used interchangeably when describing the echocardiographic phenotype. LV wall thickness greater than 12 mm (6 mm to 10 mm is normal) in the absence of hypertension should prompt suspicion for CA.34 LV thickening most often appears symmetric; however, occasionally it may exhibit asymmetric septal hypertrophy, particularly in ATTRwt-CA. In some cases, there may be a smaller subset that can actually have dynamic LV outflow obstruction similar to that seen in hypertrophic obstructive cardiomyopathy.22–24 An important echocardiographic clue that can differentiate CA from other diseases is thickening of both the LV and right ventricle (Figure 3). Septal wall thickness and LV mass index are greater in ATTR-CA compared with AL-CA.30 On average, the LV septum is around 15 mm in AL-CA and around 18 mm in ATTRwt-CA.40 Historically, the characteristic myocardial “granular sparking” or “speckling” pattern has low sensitivity and specificity.16 The left ventricle is not dilated; rather, the ventricular dimensions are usually smaller than normal. Although ejection fraction is usually preserved, cardiac output is low due to decreased ventricular volume.37 Systolic dysfunction occurs late in the disease.16 Diastolic dysfunction is universal, with a mitral inflow pattern that can range from stage I (abnormal relaxation) in early disease to stage III (restrictive filling pattern) in more advanced disease. Septal and lateral tissue Doppler velocities are very low in amyloid heart disease.41 Another echocardiographic clue to diagnosis is thickening of the heart valves, which is not seen in hypertensive heart disease or hypertrophic cardiomyopathy. Biatrial dilation is common, and there can be thickening of the interatrial septum.
Laboratory testing
N-terminal pro-b-type natriuretic peptide (NT-proBNP) is universally elevated in CA and is typically higher in AL-CA than in ATTR-CA. Troponin T or troponin I or both may be chronically elevated in CA and likely signify small-vessel ischemia. In the appropriate clinical context of a thickened ventricle and heart failure, an elevated troponin value (outside of an acute coronary syndrome) should trigger suspicion for CA. Workup for a monoclonal protein process should always be done when considering CA to rule out AL. Serum and urine protein electrophoresis are insensitive tests to detect AL and should not be relied upon as a screening test. The serum free light chain (sFLC) assay, which measures free kappa and lambda light chain levels and reports the ratio, is a sensitive test that should be measured routinely along with immunofixation of the serum and urine. In AL, sFLC will reveal an abnormal kappa-lambda ratio. An abnormally low ratio (less than 0.26) suggests a monoclonal lambda light chain process, while an abnormally high ratio (greater than 1.65) suggests a monoclonal kappa light chain process. Immunofixation will reveal an M-protein. Because light chains are excreted by the kidney, the serum levels of both kappa and lambda will be elevated in renal dysfunction, but the ratio should remain normal.16,31,34,40,42–47
Advanced noninvasive diagnostic tools for CA
Over the past decade, the ability to diagnose CA noninvasively has dramatically improved with strain imaging using 2D speckle tracking echocardiography, cardiac magnetic resonance imaging (MRI), and nuclear bone scintigraphy. These diagnostic tools have given clinicians options to pursue the diagnosis of CA without directly proceeding to endomyocardial biopsy.
Cardiac MRI. Cardiac MRI is useful for the diagnosis of CA (Figure 4B, 4C). Imaging after administration of gadolinium contrast shows a characteristic late gadolinium enhancement (LGE) pattern that is diffuse and subendocardial, and does not follow any particular coronary distribution.49 LGE can also be seen in the right ventricle and the atrial walls, and can be transmural and patchy in ATTRwt-CA. This pattern is highly sensitive (93%) and specific (70%) for CA with an overall negative predictive accuracy of 84%.50
One of the main limitations of cardiac MRI for the diagnosis of CA is the inability to give contrast in patients with reduced glomerular filtration rate. However, native T1-myocardial mapping techniques that do not require contrast show significantly increased native T1 times in CA and offer a promising alternative. Cardiac MRI parameters such as LGE, the difference in inversion time between the LV cavity and myocardium, native T1 mapping, and extracellular volume offer prognostic information. A greater than fivefold mortality increase is seen in CA patients with transmural LGE compared with those without LGE.49,50
99mTcPYP scintigraphy. 99mTcPYP, a radiotracer used in bone scans, was initially used in cardiology to quantify myocardial infarction due to its ability to localize calcium.51 Its potential utility in CA came in 1982 when diffuse myocardial 99mTcPYP uptake on cardiac radionucleotide imaging was noted in 10 patients with tissue-proven amyloidosis.52 Several subsequent studies reproduced and expanded upon this observation and revealed its diagnostic value, specifically showing that there is significant uptake in ATTR-CA and no to mild uptake in AL-CA. This offers a significant advantage over other noninvasive modalities in that it not only confirms the diagnosis of CA but differentiates ATTR-CA.16
99mTcPYP myocardial radiotracer uptake is graded by the semiquantitative visual score of cardiac retention, where grade 0 = no cardiac uptake, grade 1 = mild uptake less than bone, grade 2 = moderate uptake equal to bone, and grade 3 = high uptake greater than bone (Figure 4D).42 Additionally, quantitative analysis of heart retention can be calculated drawing circular regions of interest over the heart and mirrored on the contralateral chest wall. A heart-to-contralateral ratio greater than 1.5 is consistent with the diagnosis of ATTR-CA.53,54 In 2016, a multicenter study showed that grade 2 or 3 myocardial radiotracer uptake on bone scintigraphy in the absence of evidence of a monoclonal gammopathy was diagnostic for ATTR-CA, providing a cost-effective and non-invasive technique with a specificity and positive predictive value of 100% (confidence interval, 99.0–100%).42
Endomyocardial biopsy, right heart catheterization, and fat biopsy
Endomyocardial biopsy is essentially 100% sensitive for the diagnosis of CA.25 The main risk of pursuing endomyocardial biopsy is about a 1% risk of right ventricular perforation leading to cardiac tamponade.55 The other limitation to this approach is that not all centers are equipped to perform this procedure. Birefringence under polarized light microscopy is histopathologically diagnostic of CA; however, further subtyping by the pathologist to determine if it is AL or ATTR is absolutely crucial. Subtyping can be performed by immunohistochemistry with caution taken for misinterpretation. If there is any question of accuracy, the specimen should be sent for laser microdissection and mass spectroscopy for accurate identification of the precursor protein type (some centers routinely perform mass spectroscopy on all myocardial specimens).16,31
Right heart catheterization is nonspecific and shows restrictive hemodynamics. The right atrial waveform shows rapid x and y descents and the right ventriclular tracing may show a dip-and-plateau pattern typical of restrictive cardiomyopathy. Cardiac output can be preserved but more commonly is low.16,31
Fat pad biopsy is 60% to 80% sensitive in AL, 65% to 85% sensitive in ATTRm, and only 14% sensitive in ATTRwt, with the accuracy dependent on the operator, pathologist, and how much tissue is removed (fat pad aspirate vs biopsy specimen).56–58 Fat pad biopsy has diagnostic limitations, and a negative fat pad biopsy does not rule out amyloidosis.
DIAGNOSTIC ALGORITHM FOR CARDIAC AMYLOIDOSIS
In conjunction with laboratory tests, 99mTcPYP scan of the heart can be ordered to investigate the possibility of ATTR-CA. Grade 2 to 3 myocardial uptake in the absence of a monoclonal plasma cell process is consistent with the diagnosis of ATTR-CA. Grade 0 or 1 myocardial uptake on 99mTcPYP scan with an abnormal sFLC ratio or positive M protein on immunofixation suggests AL-CA and a bone marrow biopsy should be performed. If the patient has an abnormal sFLC ratio and grade 2 to 3 uptake on 99mTcPYP scan, the diagnosis of ATTR-CA with unrelated monoclonal gammopathy of undetermined significance should be considered. However, this would need to be reconciled by pursuing endomyocardial biopsy and accurate tissue typing. If the 99mTcPYP scan is negative, and the sFLC ratio is normal, and immunofixation is negative, a diagnosis of CA is very unlikely.16,31,40,42–47
If the diagnosis of ATTR-CA is made, genetic testing can determine the presence or absence of a mutation to differentiate ATTRm or ATTRwt, respectively. If the diagnosis of AL-CA is suggested, a bone marrow biopsy is necessary to identify and quantify the plasma cell clone.16,31,40,42–47
TREATMENT
Management of heart failure in cardiac amyloidosis
The main treatment of heart failure revolves around sodium restriction and diuretics to relieve congestion. This can prove challenging in many patients due to the narrow window between too high or too low filling pressures. A combination of loop diuretics and an aldosterone antagonist is most effective.31,34,44,45 Torsemide is preferred over furosemide due to its superior bioavailability and longer duration of action, particularly since these patients have issues with gut edema and GI absorption. Due to dependence of the cardiac output on heart rate and the tendency for orthostatic hypotension, traditional neurohormonal antagonists including beta blockers and angiotensin-converting enzyme inhibitors are neither effective nor well tolerated.60 However, in patients with atrial fibrillation, beta blockers may need to be used for rate control. Nondihydropyridine calcium channel blockers bind avidly to amyloid fibrils and are contraindicated due to risk of profound hypotension and syncope.30,31,34,44,45 Digoxin is usually avoided in CA due to concerns of increased risk of toxicity; however, it may be used with caution for rate control in atrial fibrillation given its lack of negative inotropy.61 Maintenance of normal sinus rhythm is preferable due to the importance of atrial contribution to cardiac output.
Anticoagulation in patients with atrial fibrillation and even in patients with normal sinus rhythm and poor atrial function is important due to the high risk of thromboembolic complications.62 Pacemakers are indicated for heart block or symptomatic bradycardia.63 The role of intracardiac defibrillators is controversial, but may be warranted in selected patients with AL-CA.64,65
AL treatment
Risk stratification and prognostication for AL. The most important determinant of clinical outcome in AL is the extent of cardiac involvement, as congestive heart failure and sudden cardiac death are the most common causes of death. The level of NT-proBNP and the level of either troponin T or troponin I have strong prognostic value and form the basis for the staging system in AL. Various iterations have evolved over the years, but the most widely adopted is the 4-stage system developed and validated by Mayo Clinic. This system uses a cutoff value at diagnosis for NT-proBNP greater than 1,800 ng/mL, troponin T greater than 0.025 μg/L, and the difference between kappa and lambda free light chain levels greater than 180 mg/L. Stage level increases by the number of cutoff values exceeded, with stage IV carrying a median survival of 6 months.47 Additionally, the troponin T level can help risk-stratify patients being considered for autologous stem cell transplant. In a retrospective study, troponin T greater than 0.06 μg/L was associated with increased mortality following stem cell transplant.66 Ultimately, prognosis in AL-CA is related to the hematologic response to chemotherapy.
Current treatment strategies for AL. The survival of patients with AL has improved over the years with the advent of more effective chemotherapeutic regimens that kill the underlying plasma cell clone producing the unstable light chains. The goal of treatment is to achieve a complete hematologic response with normalization of the affected light chain and sFLC ratio as well as elimination of the M protein on immunofixation.
The development of the proteasome inhibitor bortezomib has improved efficacy and survival in AL causing a faster and more complete hematologic response than prior regimens.43,67,68
The most commonly used first-line treatment consists of a 3-drug combination with the alkylating agent cyclophosphamide, the proteasome inhibitor bortezomib, and the steroid dexamethasone, which is given weekly.43 A retrospective study by Sperry et al8 showed that patients receiving an alkylating agent, bortezomib, and a steroid had the best outcomes compared with other regimens.
For patients with refractory or relapsed disease, the CD38 monoclonal antibody daratumumab can be used if patients meet myeloma criteria and has been found to be effective thus far.68,69 Newer proteasome inhibitors such as ixazomib, which is taken orally, are being studied in alternative combination regimens.70 High-dose chemotherapy with autologous stem cell transplant can be considered in patients with an acceptable cardiac risk profile and may offer more complete and durable remission than chemotherapy, although this is controversial.43,71
The cardiologist’s role in AL-CA. The hematologist directs the chemotherapy for AL but works closely with the cardiologist when there is cardiac involvement. The main role of the cardiologist is to manage volume status with diuretics, monitor for arrhythmia, and evaluate the cardiac response to treatment.43,71 Cardiac response was traditionally measured by echocardiographic changes of wall thickness, diastolic function, and ejection fraction, as well as changes in New York Heart Association (NYHA) functional class.67 However, it is uncommon to see reduction in LV wall thickness or significant improvement in ejection fraction and if it does occur, it is a slow process that usually takes more than 1 year. With the advent of longitudinal myocardial strain imaging, improvements in strain can be seen despite the lack of structural changes on echocardiogram.72 In 2012, consensus criteria defined a cardiac response as a greater than 30% reduction in NT-proBNP.73 Misfolded light chains are toxic to cardiomyocytes by causing increased oxidative stress and impairing contractility. Thus, reduction in light chain levels can lead to clinical improvement and significant reductions in NT-proBNP without changing amyloid fibril burden in the heart.
Heart transplant for patients with AL-CA. For patients who have a good hematologic response to initial chemotherapy but have limited predicted survival due to severe heart failure, heart transplant followed by autologous stem cell transplant, is a treatment strategy that can be considered. The patient must have clinically isolated severe cardiac disease, minimal amyloid burden in other organs, and a plasma cell clone that is responsive to therapy. Initial reports of heart transplant showed poor survival rates due to recurrent amyloid in the transplanted heart and progressive amyloid deposition in other organs. However, due to improved anti-plasma-cell-directed therapy and refinement in patient selection, outcomes have improved. Contemporary series of patients undergoing heart transplant followed by stem cell transplant showed that outcomes are almost comparable to heart transplant for other indications, with a 5-year survival rate of approximately 65%.74
Future therapies for AL. There is an AL amyloid-directed monoclonal antibody designed to remove amyloid fibrils from affected organs and is currently undergoing clinical trials. NEOD001, a humanized murine monoclonal antibody that targets an epitope exposed during light chain misfolding, binds to the light chain amyloid fibril and signals an immune response to clear the deposits. This agent has completed phase 1 and 2 clinical trials of 27 patients previously treated with at least 1 plasma cell-directed therapy. It showed good tolerability and achieved both renal and cardiac responses in most patients.75
A phase 2b clinical trial (NCT02632786) of patients with AL with a previous hematologic response to treatment and persistent heart dysfunction is underway and expected to be completed in January 2018. A phase 3 clinical trial (NCT02312206) of NEOD001 as an adjunct to chemotherapy is also ongoing and results are expected in February 2019.
ATTR treatment
Liver transplant for ATTRm amyloidosis for the V30M mutation that causes FAP was first described in 1990, but it has not been well validated in other mutations and is not a solution for ATTRwt.76 Significant progress has been made over the past 2 decades in the understanding of the pathophysiologyy of ATTR, paving the way for promising advancements in pharmacotherapy. Presently, there are 3 classes of pharmacologic agents, grouped by the point of disease process each strategy targets:
- Block TTR synthesis at the translational level in hepatocytes
- Stabilize the TTR tetramer to inhibit the rate-determining step of amyloidogenesis
- Disrupt and clear the ATTR amyloid fibril.77
Block TTR synthesis. TTR messenger RNA (mRNA) can be targeted by “silencers” preventing translation, thereby reducing the production TTR protein by hepatocytes. The resultant sustained reduction of plasma TTR should decrease or halt amyloid deposition by making less TTR available to dissociate and deposit in the heart and nerves. There are 2 approaches to silencing TTR mRNA translation: small interfering RNA (siRNA) and antisense oligonucleotide (ASO).77
An siRNA, packaged in a lipid nanoparticle to ensure delivery to the liver, has been designed to bind to a conserved region of TTR mRNA, degrading the mRNA and reducing TTR protein expression. One siRNA, patisiran, completed the phase 3 APOLLO clinical trial (NCT01960348) in August 2017. It is an intravenous medication that requires premedication. The APOLLO trial studied patients with neuropathic variants of ATTRm, with a primary end point of neuropathy progression over 18 months. Patisrian met the primary end point and will likely be approved by the US Food and Drug Administration (FDA) by the third quarter of 2018. Given its target of a conserved 3’ untranslated region of TTR mRNA, patisiran should theoretically yield benefits not just in FAP but to both ATTRm-CA and ATTRwt-CA.68,77
ASOs are single-stranded oligonucleotides, typically 20 nucleotides in length, that bind to mRNA and elicit enzymatic mRNA degradation and reduced protein expression. Inotersen (IONIS-TTRRx) is an ASO drug that targets a conserved region of TTR mRNA. Inotersen is administered by weekly subcutaneous injection. A phase 2/3 clinical trial (NCT01737398) completed in October 2017 studied the drug's efficacy in treating FAP, with a primary end point of neuropathy progression over 65 weeks. It met the primary end point and a subset of patients actually improved. Like patisiran, inotersen is likely to be approved by the FDA for a neuropathy indication by the third quarter of 2018 and may benefit patients with ATTRm-CA and ATTRwt-CA. More studies are needed in the ATTR cardiac population.68,77
Stabilize the TTR tetramer. The TTR tetramer has 2 thyroxine binding pockets that stabilize the structure when bound preventing dissociation. Dissociation of the tetramer, the rate limiting step for ATTR fibrillogenesis, can be reduced using pharmacologic agents that bind to the thyroxine binding pockets. Both new and repurposed agents have been found to stabilize the TTR tetramer, including diflunisal, tafamidis, tolcapone, and AG10.68,77
Diflunisal (Dolobid) is a nonacetylated salicylate nonsteroidal anti-inflammatory drug used for over 3 decades to treat arthritis and musculoskeletal pain. Unrelated to its anti-inflammatory properties, it interacts with TTR’s thyroxine binding pocket to increase the stability of the tetramer. A 2013 randomized, placebo-controlled, international multicenter trial of 130 patients with FAP demonstrated a statistically significant slowing of polyneuropathy progression; however, 67 patients (diflunisal n = 27, placebo n = 40) did not complete the study.78 A small study revealed diflunisal to be reasonably well-tolerated in ATTR-CA, and to date it is the only readily available pharmacotherapy for ATTR supported by a randomized, placebo-controlled trial.79,80 Diflunisal may be considered for off-label use in patients with ATTR-CA with relatively preserved kidney function and no increased bleeding risk, taken under the supervision of a cardiologist who will monitor for fluid retention and changes in renal function.77
Tafamidis (Vyndaqel), like diflunisal, interacts with TTR’s thyroxine binding pocket and increases tetrameric stability. A phase 2 open-label clinical trial studied the efficacy and tolerance of tafamidis in 31 patients with ATTRwt and NYHA functional class 1 and 2 followed for 1 year. Twenty-eight patients completed the study and 2 patients died. TTR stabilization at 6 weeks was achieved in 30 of 31 patients (96.8%), and success at 1 year in 25 of 28 patients (89.3%). No clinical progression occurred in 16 of 31 patients (51.5%), and tafamidis was generally well-tolerated, with diarrhea the most common side effect in 7 of 31 patients (22.6% ).81,82
A phase 3 clinical trial (NCT01994889) is studying tafamidis vs placebo in patients with ATTRm-CA or ATTRwt-CA (excluding NYHA functional class 4) with the primary outcome measure of all-cause mortality and heart failure-related hospitalizations over 30 months. This is a large trial that enrolled 446 patients and data collection is expected to be completed in February 2018.
Tolcapone, approved by the FDA for Parkinson disease, has been found to be a potent TTR stabilizer by binding both thyroxine binding pockets of the TTR tetramer simultaneously. However, tolcapone has an FDA “black box” warning due to the risk of potentially fatal acute fulminant liver failure and is not currently used in ATTR therapy.68 Nonetheless, it will likely undergo further study for ATTR.
Recruiting is underway for a phase 1 clinical trial of AG10, a potent selective TTR stabilizer (NCT03294707).68
Disruption and clearance of the ATTR amyloid fibril. Even though treatment directed at blocking TTR synthesis or stabilizing the tetramer may be effective at preventing further deposition, the residual amyloid deposits persist and continue to affect organ function. With that need in mind, several agents that disrupt the amyloid formation process further downstream have been evaluated at a basic science level and in a few small nonrandomized open-label studies.77
Doxycycline is a tetracycline antibiotic with demonstrated effectiveness in disrupting mature amyloid fibrils in mouse models.83 Tauroursodeoxycholic acid is a bile acid with the ability to disrupt prefibrillar amyloid components. This combination has been studied in patients with ATTR-CA in 2 small open-label trials, with only 1 having results published. There was no progression in NT-proBNP or wall thickness in a cohort of 7 patients who completed 12 months of treatment.84 These data are nonrandomized and hypothesis-generating, as adequate studies would need to be performed to evaluate this hypothesis further. Because there are currently no FDA-approved therapies for ATTR, patients may be offered this combination fully informed that the data are limited. (Note: ursodiol is substituted for tauroursodeoxycholic acid, which is not available in the United States.)68
Green tea extract contains the polyphenol epigallocatechin-3-gallate, which has shown the ability for fibril disruption as well as TTR stabilization, importantly using a binding site separate from the thyroxine binding pocket (utilized in diflunisal and tafamidis). A small open-label study of 19 patients with ATTR-CA of whom 14 took green tea extract for 1 year reported a reduction in intraventricular septal wall thickness at 1 year, and in 9 patients a reduction of 12.5% in LV mass measured by cardiac MRI.85
Curcumin, the active ingredient in the household spice turmeric, has displayed in vitro promise as a TTR stabilizer by binding to the thyroxine binding pocket, and as an amyloid fibril disruptor by increasing macrophage degradation activity. Although there have only been preliminary animal studies, this supplement may be promising for further study in humans with ATTR-CA.68
PRX004 is a synthetic antibody designed to bind to non-native misfolded forms of TTR with the goal of potentially preventing deposition and promoting clearance of TTR aggregates.86 A phase 1 open-label escalation trial (NCT03336580) in 36 patients with ATTRm is planned and, hopefully, will pave the way for further study.
Heart transplant for patients with ATTR-CA. Patients with ATTRwt-CA who are young enough to undergo heart transplant have displayed favorable outcomes given that it causes clinically isolated heart disease and is an indolent process that should not affect the transplanted heart over the average life span of the allograft. Patients with the ATTRm mutation V122I have been treated with heart transplant alone, with the thought process that again, due to the indolent nature of amyloid deposition, concomitant liver transplant may not be needed.74 Thus far, 6 patients with this mutation have undergone successful transplant at our institution with heart alone, 1 of whom is 9 years posttransplant without any recurrent amyloid in the allograft. Patients with the T60A mutation that causes both polyneuropathy and cardiomyopathy require combined heart and liver transplant.74
Are there other therapies being studied to clear amyloid deposits and reverse organ dysfunction?
Extracellular deposits of amyloid fibrils, regardless of precursor protein, contain common elements such as calcium, glycosaminoglycans, and an SAP component. SAP stabilizes amyloid fibrils and makes them resistant to degradation. A monoclonal immunoglobulin G1 anti-SAP antibody has been designed to target the ubiquitous SAP component, signaling an immune response that leads to macrophage mediated clearance of amyloid fibrils, regardless of the type. Treatment with this approach was studied in a pilot trial of 16 patients mostly with AL, and within a 6-week period some of the patients had dramatic reversal of liver amyloid deposition.2 This has paved the way for a phase 2 open-label trial to be performed in patients with AL-CA and ATTR-CA (NCT03044353) and plans for a randomized phase 3 trial in CA are in discussion. There is optimism that this therapy may achieve the holy grail of removing amyloid rather than just preventing further deposition.
CONCLUSION
The diagnosis of CA requires a high index of suspicion. The diagnostic tools have improved due to the availability of modern imaging techniques, and the advent of measuring the sFLC assay along with immunofixation of the serum and urine. The prognosis for patients with AL-CA used to be dismal, with very poor survival rates. The current treatment strategies that include proteasome inhibitors have significantly improved survival, emphasizing the importance of early diagnosis and prompt initiation of therapy. Monoclonal antibodies against plasma cells (daratumumab) and light chain amyloid deposits (NEOD001) have the potential to further improve outcomes. The diagnosis of ATTR-CA used to be a futile academic pursuit given the lack of available therapies. However, there are several new FDA-approved agents on the horizon, including TTR gene silencers and stabilizers. CA is no longer considered to be rare and hopeless. Rather, it is more common than previously recognized and even more treatable.
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med 2003; 349:583–596.
- Richards DB, Cookson LM, Berges AC, et al. Therapeutic clearance of amyloid by antibodies to serum amyloid P component. N Engl J Med 2015; 373:1106–1114.
- Sipe JD, Benson MD, Buxbaum JN, et al. Nomenclature 2014: amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 2014; 21:221–224.
- Maleszewski JJ. Cardiac amyloidosis: pathology, nomenclature, and typing. Cardiovasc Pathol 2015; 24:343–350.
- Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood 1992; 79:1817–1822.
- Krsnik I, Cabero M, Morillo D, et al. Light chain amyloidosis: Experience in a tertiary hospital: 2005-2013. Rev Clin Esp 2015; 215:1–8.
- Dubrey SW, Cha K, Anderson J, et al. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM 1998; 91:141–157.
- Sperry BW, Ikram A, Hachamovitch R, et al. Efficacy of chemotherapy for light-chain amyloidosis in patients presenting with symptomatic heart failure. J Am Coll Cardiol 2016; 67:2941–2948.
- Gillmore JD, Damy T, Fontana M, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J 2017; Oct 18. [Epub ahead of print] doi:10.1093/eurheartj/ehx589.
- Tanskanen M, Peuralinna T, Polvikoski T, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med 2008; 40:232–239.
- González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015; 36:2585–2594.
- Longhi S, Lorenzini M, Gagliardi C, et al. Coexistence of degenerative aortic stenosis and wild-type transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging 2016;9:325–327.
- Grogan M, Scott CG, Kyle RA, et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol 2016; 68:1014–1020.
- Gioeva Z, Urban P, Meliss RR, et al. ATTR amyloid in the carpal tunnel ligament is frequently of wildtype transthyretin origin. Amyloid 2013; 20:1–6.
- Yanagisawa A, Ueda M, Sueyoshi T, et al. Amyloid deposits derived from transthyretin in the ligamentum flavum as related to lumbar spinal canal stenosis. Mod Pathol 2015; 28:201–207.
- Siddiqi OK, Ruberg FL. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med 2017; Jul 13. [Epub ahead of print] doi:10.1016/j.tcm.2017.07.004.
- Nakagawa M, Sekijima Y, Yazaki M, et al. Carpal tunnel syndrome: A common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid 2016; 23:58–63.
- Sekijima Y, Yoshida K, Tokuda T, Ikeda S-I. Familial Transthyretin Amyloidosis. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. Seattle, WA: University of Washington, Seattle; 1993–2017. Available at: https://www-ncbi-nlm-nih-gov.ccmain.ohionet.org/books/NBK1194. Updated January 26, 2012. Accessed November 16, 2017.
- Buxbaum J, Jacobson DR, Tagoe C, et al. Transthyretin V122I in African Americans with congestive heart failure. J Am Coll Cardiol 2006; 47:1724–1725.
- Ruberg FL, Maurer MS, Judge DP, et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: The Transthyretin Amyloidosis Cardiac Study (TRACS). Am Heart J 2012; 164:222–228.e1.
- Wright JR, Calkins E. Amyloid in the aged heart: frequency and clinical significance. J Am Geriatr Soc 1975; 23:97–103.
- Vermeer AMC, Janssen A, Boorsma PC, Mannens MMAM, Wilde AAM, Christiaans I. Transthyretin amyloidosis: a phenocopy of hypertrophic cardiomyopathy. Amyloid 2017; 24:87–91.
- Velazquez-Ceceña JL, Lubell DL, Nagajothi N, Al-Masri H, Siddiqui M, Khosla S. Syncope cardiomyopathy in a patient with primary AL-type amyloid heart disease. Tex Heart Inst J 2009; 36:50–54.
- Stegman BM, Kwon D, Rodriguez ER, Hanna M, Cho L. Left ventricular hypertrophy in a runner: things are not always what they seem. Circulation 2014; 130:590–592.
- Ardehali H, Qasim A, Cappola T, et al. Endomyocardial biopsy plays a role in diagnosing patients with unexplained cardiomyopathy. Am Heart J 2004; 147:919–923.
- Berghoff M, Kathpal M, Khan F, Skinner M, Falk R, Freeman R. Endothelial dysfunction precedes C-fiber abnormalities in primary (AL) amyloidosis. Ann Neurol 2003; 53:725–730.
- Khan MF, Falk RH. Amyloidosis. Postgrad Med J 2001; 77:686–693.
- Martinez-Naharro A, Treibel TA, Abdel-Gadir A, et al. Magnetic resonance in transthyretin cardiac amyloidosis. J Am Coll Cardiol 2017; 70:466–477.
- Longhi S, Quarta CC, Milandri A, et al. Atrial fibrillation in amyloidotic cardiomyopathy: prevalence, incidence, risk factors and prognostic role. Amyloid 2015; 22:147–155.
- Sperry BW, Vranian MN, Hachamovitch R, et al. Are classic predictors of voltage valid in cardiac amyloidosis? A contemporary analysis of electrocardiographic findings. Int J Cardiol 2016; 214:477–481.
- Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation 2005; 112:2047–2060.
- Berk JL, Keane J, Seldin DC, et al. Persistent pleural effusions in primary systemic amyloidosis: etiology and prognosis. Chest 2003; 124:969–977.
- Miani D, Rocco M, Alberti E, Spedicato L, Fioretti PM. Amyloidosis of epicardial and intramural coronary arteries as an unusual cause of myocardial infarction and refractory angina pectoris. Ital Heart J 2002; 3:479–482.
- Mesquita ET, Jorge AJL, Souza CV Junior, Andrade TR. Cardiac amyloidosis and its new clinical phenotype: heart failure with preserved ejection fraction. Arg Bras Cardiol 2017; 109:71–80.
- Swanton RH, Brooksby IAB, Davies MJ, Coltart DJ, Jenkins BS, Webb-Peploe MM. Systolic and diastolic ventricular function in cardiac amyloidosis: studies in six cases diagnosed with endomyocardial biopsy. Am J Cardiol 1977; 39:658–664.
- Tyberg TI, Goodyer AVN, Hurst VW 3rd, Alexander J, Langou RA. Left ventricular filling in differentiating restrictive amyloid cardiomyopathy and constrictive pericarditis. Am J Cardiol 1981; 47:791–796.
- Sperry BW, Jones BM, Vranian MN, Hanna M, Jaber WA. Recognizing transthyretin cardiac amyloidosis in patients with aortic stenosis: impact on prognosis. JACC Cardiovasc Imaging 2016; 9:904–906.
- Cyrille NB, Goldsmith J, Alvarez J, Maurer MS. Prevalence and prognostic significance of low QRS voltage among the three main types of cardiac amyloidosis. Am J Cardiol 2014; 114:1089–1093.
- Hongo M, Yamamoto H, Kohda T, et al. Comparison of electrocardiographic findings in patients with AL (primary) amyloidosis and in familial amyloid polyneuropathy and anginal pain and their relation to histopathologic findings. Am J Cardiol 2000; 85:849–853.
- Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation 2017; 135:1357–1377.
- Abdalla I, Murray RD, Lee JC, Stewart WJ, Tajik AJ, Klein AL. Duration of pulmonary venous atrial reversal flow velocity and mitral inflow a wave: new measure of severity of cardiac amyloidosis. J Am Soc Echocardiogr 1998; 11:1125–1133.
- Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016; 133:2404–2412.
- Gertz MA. Immunoglobulin light chain amyloidosis: 2016 update on diagnosis, prognosis, and treatment. Am J Hematol 2016; 91:947–956.
- Patel KS, Hawkins PN. Cardiac amyloidosis: where are we today? J Intern Med 2015; 278:126–144.
- Ritts AJ, Cornell RF, Swiger K, Singh J, Goodman S, Lenihan DJ. Current concepts of cardiac amyloidosis: diagnosis, clinical management, and the need for collaboration. Heart Fail Clin 2017; 13:409–416.
- Sperry BW, Vranian MN, Hachamovitch R, et al. Subtype-specific interactions and prognosis in cardiac amyloidosis. J Am Heart Assoc 2016; 5:e002877.
- Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol 2012; 30:989–995.
- Phelan D, Collier P, Thavendiranathan P, et al. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart 2012; 98:1442–1448.
- Patel AR, Kramer CM. Role of cardiac magnetic resonance in the diagnosis and prognosis of nonischemic cardiomyopathy. JACC Cardiovasc Imaging 2017; 10(10 Pt A):1180–1193.
- White JA, Kim HW, Shah D, et al. CMR imaging with rapid visual T1 assessment predicts mortality in patients suspected of cardiac amyloidosis. JACC Cardiovasc Imaging 2014; 7:143–156.
- Parkey RW, Bonte FJ, Meyer SL, et al. A new method for radionuclide imaging of acute myocardial infarction in humans. Circulation 1974; 50:540–546.
- Wizenberg TA, Muz J, Sohn YH, Samlowski W, Weissler AM. Value of positive myocardial technetium-99m-pyrophosphate scintigraphy in the noninvasive diagnosis of cardiac amyloidosis. Am Heart J 1982; 103(4 Pt 1):468–473.
- Bokhari S, Castaño A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. 99mTc-Pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging 2013; 6:195–201.
- Vranian MN, Sperry BW, Hanna M, et al. Technetium pyrophosphate uptake in transthyretin cardiac amyloidosis: associations with echocardiographic disease severity and outcomes. J Nucl Cardiol 2017; Jan 3. [Epub ahead of print] doi: 10.1007/s12350-016-0768-9.
- Bennett MK, Gilotra NA, Harrington C, et al. Evaluation of the role of endomyocardial biopsy in 851 patients with unexplained heart failure from 2000-2009. Circ Heart Fail 2013; 6:676–684.
- Garcia Y, Collins AB, Stone JR. Abdominal fat pad excisional biopsy for the diagnosis and typing of systemic amyloidosis. Hum Pathol 2017; Nov 10. [Epub ahead of print] doi:10.1016/j.humpath.2017.11.001.
- Quarta CC, Gonzalez-Lopez E, Gilbertson JA, et al. Diagnostic sensitivity of abdominal fat aspiration in cardiac amyloidosis. Eur Heart J 2017; 38:1905–1908.
- Fine NM, Arruda-Olson AM, Dispenzieri A, et al. Yield of noncardiac biopsy for the diagnosis of transthyretin cardiac amyloidosis. Am J Cardiol 2014; 113:1723–1727.
- Katzmann JA, Abraham RS, Dispenzieri A, Lust JA, Kyle RA. Diagnostic performance of quantitative kappa and lambda free light chain assays in clinical practice. Clin Chem 2005; 51:878–781.
- Malha L, Mann SJ. Loop diuretics in the treatment of hypertension. Curr Hypertens Rep 2016; 18:27.
- Rubinow A, Skinner M, Cohen AS. Digoxin sensitivity in amyloid cardiomyopathy. Circulation 1981; 63:1285–1288.
- Feng D, Syed IS, Martinez M, et al. Intracardiac thrombosis and anticoagulation therapy in cardiac amyloidosis. Circulation 2009; 119:2490–2497.
- Castaño A, Drachman BM, Judge D, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev 2015; 20:163–178.
- Hamon D, Algalarrondo V, Gandjbakhch E, et al. Outcome and incidence of appropriate implantable cardioverter-defibrillator therapy in patients with cardiac amyloidosis. Int J Cardiol 2016; 222:562–568.
- Patel KS, Hawkins PN, Whelan CJ, Gillmore JD. Life-saving implantable cardioverter defibrillator therapy in cardiac AL amyloidosis. BMJ Case Rep 2014; Dec 22. doi:10.1136/bcr-2014-206600.
- Gertz M, Lacy M, Dispenzieri A, et al. Troponin T level as an exclusion criterion for stem cell transplantation in light-chain amyloidosis. Leuk Lymphoma 2008; 49:36–41.
- Vranian MN, Sperry BW, Valent J, Hanna M. Emerging advances in the management of cardiac amyloidosis. Curr Cardiol Rep 2015; 17:100.
- Alexander KM, Singh A, Falk RH. Novel pharmacotherapies for cardiac amyloidosis. Pharmacol Ther 2017; 180:129–138.
- Sher T, Fenton B, Akhtar A, Gertz MA. First report of safety and efficacy of daratumumab in 2 cases of advanced immunoglobulin light chain amyloidosis. Blood 2016; 128:1987–1989.
- Sanchorawala V, Palladini G, Kukreti V, et al. A phase 1/2 study of the oral proteasome inhibitor ixazomib in relapsed or refractory AL amyloidosis. Blood 2017; 130:597–605.
- Zumbo G, Sadeghi-Alavijeh O, Hawkins PN, Fontana M. New and developing therapies for AL amyloidosis. Expert Opin Pharmacother 2017; 18:139–149.
- Koyama J, Falk RH. Prognostic significance of strain Doppler imaging in light-chain amyloidosis. JACC Cardiovasc Imaging 2010; 3:333–342.
- Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol 2012; 30:4541–4549.
- Sousa M, Monohan G, Rajagopalan N, Grigorian A. Heart transplantation in cardiac amyloidosis. Heart Fail Rev 2017; 22:317–327.
- Gertz MA, Landau H, Comenzo RL, et al. First-in-human phase I/II study of NEOD001 in patients with light chain amyloidosis and persistent organ dysfunction. J Clin Oncol 2016; 34:1097–1103.
- Holmgren G, Steen L, Ekstedt J, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet 1991; 40:242–246.
- Hanna M. Novel drugs targeting transthyretin amyloidosis. Curr Heart Fail Rep 2014; 11:50–57.
- Berk JL, Suhr OB, Obici L, et al; Diflunisal Trial Consortium. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013; 310:2658–2667.
- Castaño A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail 2012; 18:315–319.
- Sekijima Y, Tojo K, Morita H, Koyama J, Ikeda S-I. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid 2015; 22:79–83.
- Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012; 79:785–792.
- Maurer MS, Grogan DR, Judge DP, et al. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ Heart Fail 2015; 8:519–526.
- Cardoso I, Martins D, Ribeiro T, Merlini G, Saraiva MJ. Synergy of combined doxycycline/TUDCA treatment in lowering transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med 2010; 8:74.
- Obici L, Cortese A, Lozza A, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: A phase II study. Amyloid 2012; 19(suppl 1):34–36.
- Kristen AV, Lehrke S, Buss S, et al. Green tea halts progression of cardiac transthyretin amyloidosis: An observational report. Clin Res Cardiol 2012; 101:805–813.
- Higaki JN, Chakrabartty A, Galant NJ, et al. Novel conformation-specific monoclonal antibodies against amyloidogenic forms of forms of transthyretin. Amyloid 2016; 23:86–97.
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med 2003; 349:583–596.
- Richards DB, Cookson LM, Berges AC, et al. Therapeutic clearance of amyloid by antibodies to serum amyloid P component. N Engl J Med 2015; 373:1106–1114.
- Sipe JD, Benson MD, Buxbaum JN, et al. Nomenclature 2014: amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 2014; 21:221–224.
- Maleszewski JJ. Cardiac amyloidosis: pathology, nomenclature, and typing. Cardiovasc Pathol 2015; 24:343–350.
- Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood 1992; 79:1817–1822.
- Krsnik I, Cabero M, Morillo D, et al. Light chain amyloidosis: Experience in a tertiary hospital: 2005-2013. Rev Clin Esp 2015; 215:1–8.
- Dubrey SW, Cha K, Anderson J, et al. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM 1998; 91:141–157.
- Sperry BW, Ikram A, Hachamovitch R, et al. Efficacy of chemotherapy for light-chain amyloidosis in patients presenting with symptomatic heart failure. J Am Coll Cardiol 2016; 67:2941–2948.
- Gillmore JD, Damy T, Fontana M, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J 2017; Oct 18. [Epub ahead of print] doi:10.1093/eurheartj/ehx589.
- Tanskanen M, Peuralinna T, Polvikoski T, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med 2008; 40:232–239.
- González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J 2015; 36:2585–2594.
- Longhi S, Lorenzini M, Gagliardi C, et al. Coexistence of degenerative aortic stenosis and wild-type transthyretin-related cardiac amyloidosis. JACC Cardiovasc Imaging 2016;9:325–327.
- Grogan M, Scott CG, Kyle RA, et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol 2016; 68:1014–1020.
- Gioeva Z, Urban P, Meliss RR, et al. ATTR amyloid in the carpal tunnel ligament is frequently of wildtype transthyretin origin. Amyloid 2013; 20:1–6.
- Yanagisawa A, Ueda M, Sueyoshi T, et al. Amyloid deposits derived from transthyretin in the ligamentum flavum as related to lumbar spinal canal stenosis. Mod Pathol 2015; 28:201–207.
- Siddiqi OK, Ruberg FL. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med 2017; Jul 13. [Epub ahead of print] doi:10.1016/j.tcm.2017.07.004.
- Nakagawa M, Sekijima Y, Yazaki M, et al. Carpal tunnel syndrome: A common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid 2016; 23:58–63.
- Sekijima Y, Yoshida K, Tokuda T, Ikeda S-I. Familial Transthyretin Amyloidosis. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. Seattle, WA: University of Washington, Seattle; 1993–2017. Available at: https://www-ncbi-nlm-nih-gov.ccmain.ohionet.org/books/NBK1194. Updated January 26, 2012. Accessed November 16, 2017.
- Buxbaum J, Jacobson DR, Tagoe C, et al. Transthyretin V122I in African Americans with congestive heart failure. J Am Coll Cardiol 2006; 47:1724–1725.
- Ruberg FL, Maurer MS, Judge DP, et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: The Transthyretin Amyloidosis Cardiac Study (TRACS). Am Heart J 2012; 164:222–228.e1.
- Wright JR, Calkins E. Amyloid in the aged heart: frequency and clinical significance. J Am Geriatr Soc 1975; 23:97–103.
- Vermeer AMC, Janssen A, Boorsma PC, Mannens MMAM, Wilde AAM, Christiaans I. Transthyretin amyloidosis: a phenocopy of hypertrophic cardiomyopathy. Amyloid 2017; 24:87–91.
- Velazquez-Ceceña JL, Lubell DL, Nagajothi N, Al-Masri H, Siddiqui M, Khosla S. Syncope cardiomyopathy in a patient with primary AL-type amyloid heart disease. Tex Heart Inst J 2009; 36:50–54.
- Stegman BM, Kwon D, Rodriguez ER, Hanna M, Cho L. Left ventricular hypertrophy in a runner: things are not always what they seem. Circulation 2014; 130:590–592.
- Ardehali H, Qasim A, Cappola T, et al. Endomyocardial biopsy plays a role in diagnosing patients with unexplained cardiomyopathy. Am Heart J 2004; 147:919–923.
- Berghoff M, Kathpal M, Khan F, Skinner M, Falk R, Freeman R. Endothelial dysfunction precedes C-fiber abnormalities in primary (AL) amyloidosis. Ann Neurol 2003; 53:725–730.
- Khan MF, Falk RH. Amyloidosis. Postgrad Med J 2001; 77:686–693.
- Martinez-Naharro A, Treibel TA, Abdel-Gadir A, et al. Magnetic resonance in transthyretin cardiac amyloidosis. J Am Coll Cardiol 2017; 70:466–477.
- Longhi S, Quarta CC, Milandri A, et al. Atrial fibrillation in amyloidotic cardiomyopathy: prevalence, incidence, risk factors and prognostic role. Amyloid 2015; 22:147–155.
- Sperry BW, Vranian MN, Hachamovitch R, et al. Are classic predictors of voltage valid in cardiac amyloidosis? A contemporary analysis of electrocardiographic findings. Int J Cardiol 2016; 214:477–481.
- Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation 2005; 112:2047–2060.
- Berk JL, Keane J, Seldin DC, et al. Persistent pleural effusions in primary systemic amyloidosis: etiology and prognosis. Chest 2003; 124:969–977.
- Miani D, Rocco M, Alberti E, Spedicato L, Fioretti PM. Amyloidosis of epicardial and intramural coronary arteries as an unusual cause of myocardial infarction and refractory angina pectoris. Ital Heart J 2002; 3:479–482.
- Mesquita ET, Jorge AJL, Souza CV Junior, Andrade TR. Cardiac amyloidosis and its new clinical phenotype: heart failure with preserved ejection fraction. Arg Bras Cardiol 2017; 109:71–80.
- Swanton RH, Brooksby IAB, Davies MJ, Coltart DJ, Jenkins BS, Webb-Peploe MM. Systolic and diastolic ventricular function in cardiac amyloidosis: studies in six cases diagnosed with endomyocardial biopsy. Am J Cardiol 1977; 39:658–664.
- Tyberg TI, Goodyer AVN, Hurst VW 3rd, Alexander J, Langou RA. Left ventricular filling in differentiating restrictive amyloid cardiomyopathy and constrictive pericarditis. Am J Cardiol 1981; 47:791–796.
- Sperry BW, Jones BM, Vranian MN, Hanna M, Jaber WA. Recognizing transthyretin cardiac amyloidosis in patients with aortic stenosis: impact on prognosis. JACC Cardiovasc Imaging 2016; 9:904–906.
- Cyrille NB, Goldsmith J, Alvarez J, Maurer MS. Prevalence and prognostic significance of low QRS voltage among the three main types of cardiac amyloidosis. Am J Cardiol 2014; 114:1089–1093.
- Hongo M, Yamamoto H, Kohda T, et al. Comparison of electrocardiographic findings in patients with AL (primary) amyloidosis and in familial amyloid polyneuropathy and anginal pain and their relation to histopathologic findings. Am J Cardiol 2000; 85:849–853.
- Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation 2017; 135:1357–1377.
- Abdalla I, Murray RD, Lee JC, Stewart WJ, Tajik AJ, Klein AL. Duration of pulmonary venous atrial reversal flow velocity and mitral inflow a wave: new measure of severity of cardiac amyloidosis. J Am Soc Echocardiogr 1998; 11:1125–1133.
- Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016; 133:2404–2412.
- Gertz MA. Immunoglobulin light chain amyloidosis: 2016 update on diagnosis, prognosis, and treatment. Am J Hematol 2016; 91:947–956.
- Patel KS, Hawkins PN. Cardiac amyloidosis: where are we today? J Intern Med 2015; 278:126–144.
- Ritts AJ, Cornell RF, Swiger K, Singh J, Goodman S, Lenihan DJ. Current concepts of cardiac amyloidosis: diagnosis, clinical management, and the need for collaboration. Heart Fail Clin 2017; 13:409–416.
- Sperry BW, Vranian MN, Hachamovitch R, et al. Subtype-specific interactions and prognosis in cardiac amyloidosis. J Am Heart Assoc 2016; 5:e002877.
- Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol 2012; 30:989–995.
- Phelan D, Collier P, Thavendiranathan P, et al. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart 2012; 98:1442–1448.
- Patel AR, Kramer CM. Role of cardiac magnetic resonance in the diagnosis and prognosis of nonischemic cardiomyopathy. JACC Cardiovasc Imaging 2017; 10(10 Pt A):1180–1193.
- White JA, Kim HW, Shah D, et al. CMR imaging with rapid visual T1 assessment predicts mortality in patients suspected of cardiac amyloidosis. JACC Cardiovasc Imaging 2014; 7:143–156.
- Parkey RW, Bonte FJ, Meyer SL, et al. A new method for radionuclide imaging of acute myocardial infarction in humans. Circulation 1974; 50:540–546.
- Wizenberg TA, Muz J, Sohn YH, Samlowski W, Weissler AM. Value of positive myocardial technetium-99m-pyrophosphate scintigraphy in the noninvasive diagnosis of cardiac amyloidosis. Am Heart J 1982; 103(4 Pt 1):468–473.
- Bokhari S, Castaño A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. 99mTc-Pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging 2013; 6:195–201.
- Vranian MN, Sperry BW, Hanna M, et al. Technetium pyrophosphate uptake in transthyretin cardiac amyloidosis: associations with echocardiographic disease severity and outcomes. J Nucl Cardiol 2017; Jan 3. [Epub ahead of print] doi: 10.1007/s12350-016-0768-9.
- Bennett MK, Gilotra NA, Harrington C, et al. Evaluation of the role of endomyocardial biopsy in 851 patients with unexplained heart failure from 2000-2009. Circ Heart Fail 2013; 6:676–684.
- Garcia Y, Collins AB, Stone JR. Abdominal fat pad excisional biopsy for the diagnosis and typing of systemic amyloidosis. Hum Pathol 2017; Nov 10. [Epub ahead of print] doi:10.1016/j.humpath.2017.11.001.
- Quarta CC, Gonzalez-Lopez E, Gilbertson JA, et al. Diagnostic sensitivity of abdominal fat aspiration in cardiac amyloidosis. Eur Heart J 2017; 38:1905–1908.
- Fine NM, Arruda-Olson AM, Dispenzieri A, et al. Yield of noncardiac biopsy for the diagnosis of transthyretin cardiac amyloidosis. Am J Cardiol 2014; 113:1723–1727.
- Katzmann JA, Abraham RS, Dispenzieri A, Lust JA, Kyle RA. Diagnostic performance of quantitative kappa and lambda free light chain assays in clinical practice. Clin Chem 2005; 51:878–781.
- Malha L, Mann SJ. Loop diuretics in the treatment of hypertension. Curr Hypertens Rep 2016; 18:27.
- Rubinow A, Skinner M, Cohen AS. Digoxin sensitivity in amyloid cardiomyopathy. Circulation 1981; 63:1285–1288.
- Feng D, Syed IS, Martinez M, et al. Intracardiac thrombosis and anticoagulation therapy in cardiac amyloidosis. Circulation 2009; 119:2490–2497.
- Castaño A, Drachman BM, Judge D, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev 2015; 20:163–178.
- Hamon D, Algalarrondo V, Gandjbakhch E, et al. Outcome and incidence of appropriate implantable cardioverter-defibrillator therapy in patients with cardiac amyloidosis. Int J Cardiol 2016; 222:562–568.
- Patel KS, Hawkins PN, Whelan CJ, Gillmore JD. Life-saving implantable cardioverter defibrillator therapy in cardiac AL amyloidosis. BMJ Case Rep 2014; Dec 22. doi:10.1136/bcr-2014-206600.
- Gertz M, Lacy M, Dispenzieri A, et al. Troponin T level as an exclusion criterion for stem cell transplantation in light-chain amyloidosis. Leuk Lymphoma 2008; 49:36–41.
- Vranian MN, Sperry BW, Valent J, Hanna M. Emerging advances in the management of cardiac amyloidosis. Curr Cardiol Rep 2015; 17:100.
- Alexander KM, Singh A, Falk RH. Novel pharmacotherapies for cardiac amyloidosis. Pharmacol Ther 2017; 180:129–138.
- Sher T, Fenton B, Akhtar A, Gertz MA. First report of safety and efficacy of daratumumab in 2 cases of advanced immunoglobulin light chain amyloidosis. Blood 2016; 128:1987–1989.
- Sanchorawala V, Palladini G, Kukreti V, et al. A phase 1/2 study of the oral proteasome inhibitor ixazomib in relapsed or refractory AL amyloidosis. Blood 2017; 130:597–605.
- Zumbo G, Sadeghi-Alavijeh O, Hawkins PN, Fontana M. New and developing therapies for AL amyloidosis. Expert Opin Pharmacother 2017; 18:139–149.
- Koyama J, Falk RH. Prognostic significance of strain Doppler imaging in light-chain amyloidosis. JACC Cardiovasc Imaging 2010; 3:333–342.
- Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol 2012; 30:4541–4549.
- Sousa M, Monohan G, Rajagopalan N, Grigorian A. Heart transplantation in cardiac amyloidosis. Heart Fail Rev 2017; 22:317–327.
- Gertz MA, Landau H, Comenzo RL, et al. First-in-human phase I/II study of NEOD001 in patients with light chain amyloidosis and persistent organ dysfunction. J Clin Oncol 2016; 34:1097–1103.
- Holmgren G, Steen L, Ekstedt J, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet 1991; 40:242–246.
- Hanna M. Novel drugs targeting transthyretin amyloidosis. Curr Heart Fail Rep 2014; 11:50–57.
- Berk JL, Suhr OB, Obici L, et al; Diflunisal Trial Consortium. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013; 310:2658–2667.
- Castaño A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail 2012; 18:315–319.
- Sekijima Y, Tojo K, Morita H, Koyama J, Ikeda S-I. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid 2015; 22:79–83.
- Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012; 79:785–792.
- Maurer MS, Grogan DR, Judge DP, et al. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ Heart Fail 2015; 8:519–526.
- Cardoso I, Martins D, Ribeiro T, Merlini G, Saraiva MJ. Synergy of combined doxycycline/TUDCA treatment in lowering transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med 2010; 8:74.
- Obici L, Cortese A, Lozza A, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: A phase II study. Amyloid 2012; 19(suppl 1):34–36.
- Kristen AV, Lehrke S, Buss S, et al. Green tea halts progression of cardiac transthyretin amyloidosis: An observational report. Clin Res Cardiol 2012; 101:805–813.
- Higaki JN, Chakrabartty A, Galant NJ, et al. Novel conformation-specific monoclonal antibodies against amyloidogenic forms of forms of transthyretin. Amyloid 2016; 23:86–97.
KEY POINTS
- AL and ATTR are the 2 main types of amyloidosis that affect the heart.
- Serum and urine protein electrophoresis are inadequate laboratory tests to screen for AL given low sensitivity, and should be replaced by the serum free light chain assay as well as immunofixation of the serum and urine.
- AL cardiac amyloidosis (AL-CA) requires timely diagnosis and referral to hematology due to high mortality without prompt treatment.
- 99mTechnetium pyrophosphate bone scintigraphy is an affordable, noninvasive tool that has revolutionized the diagnosis of ATTR cardiac amyloidosis (ATTR-CA).
- The US Food and Drug Administration will likely approve new therapies for ATTR in late 2018.