User login
A 69-year-old woman was admitted to the hospital with double vision, weakness in the lower extremities, sensory loss, pain, and falls. Her symptoms started with sudden onset of horizontal diplopia 6 weeks before, followed by gradually worsening lower-extremity weakness, as well as ataxia and patchy and bilateral radicular burning leg pain more pronounced on the right. Her medical history included narcolepsy, obstructive sleep apnea, hypertension, hyperlipidemia, and bilateral knee replacements for osteoarthritis.
Neurologic examination showed inability to abduct the right eye, bilateral hip flexion weakness, decreased pinprick response, decreased proprioception, and diminished muscle stretch reflexes in the lower extremities. Magnetic resonance imaging (MRI) of the brain without contrast and magnetic resonance angiography of the brain and carotid arteries showed no evidence of acute stroke. No abnormalities were noted on electrocardiography and echocardiography.
A diagnosis of idiopathic peripheral neuropathy was made, and outpatient physical therapy was recommended. Over the subsequent 2 weeks, her condition declined to the point where she needed a walker. She continued to have worsening leg weakness with falls, prompting hospital readmission.
INITIAL EVALUATION
In addition to her diplopia and weakness, she said she had lost 15 pounds since the onset of symptoms and had experienced symptoms suggesting urinary retention.
Physical examination
Her temperature was 37°C (98.6°F), heart rate 79 beats per minute, blood pressure 117/86 mm Hg, respiratory rate 14 breaths per minute, and oxygen saturation 98% on room air. Examination of the head, neck, heart, lung, abdomen, lymph nodes, and extremities yielded nothing remarkable except for chronic venous changes in the lower extremities.
The neurologic examination showed incomplete lateral gaze bilaterally (cranial nerve VI dysfunction). Strength in the upper extremities was normal. In the legs, the Medical Research Council scale score for proximal muscle strength was 2 to 3 out of 5, and for distal muscles 3 to 4 out of 5, with the right side worse than the left and flexors and extensors affected equally. Muscle stretch reflexes were absent in both lower extremities and the left upper extremity, but intact in the right upper extremity. No abnormal corticospinal tract reflexes were elicited.
Sensory testing revealed diminished pin-prick perception in a length-dependent fashion in the lower extremities, reduced 50% compared with the hands. Gait could not be assessed due to weakness.
Initial laboratory testing
Results of initial laboratory tests—complete blood cell count, complete metabolic panel, erythrocyte sedimentation rate, C-reactive protein, thyroid-stimulating hormone, and hemoglobin A1c—were unremarkable.
FURTHER EVALUATION AND DIFFERENTIAL DIAGNOSIS
1. Which of the following is the most likely diagnosis at this point?
- Cerebral infarction
- Guillain-Barré syndrome
- Progressive polyneuropathy
- Transverse myelitis
- Polyradiculopathy
In the absence of definitive diagnostic tests, all of the above options were considered in the differential diagnosis for this patient.
Cerebral infarction
Although acute-onset diplopia can be explained by brainstem stroke involving cranial nerve nuclei or their projections, the onset of diplopia with progressive bilateral lower-extremity weakness makes stroke unlikely. Flaccid paralysis, areflexia of the lower extremities, and sensory involvement can also be caused by acute anterior spinal artery occlusion leading to spinal cord infarction; however, the deficits are usually maximal at onset.
Guillain-Barré syndrome
The combination of acute-subacute progressive ascending weakness, sensory involvement, and diminished or absent reflexes is typical of Guillain-Barré syndrome. Cranial nerve involvement can overlap with the more typical features of the syndrome. However, most patients reach the nadir of their disease by 4 weeks after initial symptom onset, even without treatment.1 This patient’s condition continued to worsen over 8 weeks. In addition, the asymmetric lower-extremity weakness and sparing of the arms are atypical for Guillain-Barré syndrome.
Given the progression of symptoms, chronic inflammatory demyelinating polyneuropathy is also a consideration, typically presenting as a relapsing or progressive neuropathy in proximal and distal muscles and worsening over at least an 8-week period.2
The initial workup for Guillain-Barré syndrome or chronic inflammatory demyelinating polyneuropathy includes lumbar puncture to assess for albuminocytologic dissociation (elevated protein with normal white blood cell count) in cerebrospinal fluid (CSF), and electromyography (EMG) to assess for neurophysiologic evidence of peripheral nerve demyelination. In Miller-Fisher syndrome, a rare variant of Guillain-Barré syndrome characterized by ataxia, ophthalmoparesis, and areflexia, serum ganglioside antibodies to GQ1b are found in over 90% of patients.3,4 Although MRI of the spine is not necessary to diagnose Guillain-Barré syndrome, it is often done to exclude other causes of lower-extremity weakness such as spinal cord or cauda equina compression that would require urgent neurosurgical consultation. MRI can support the diagnosis of Guillain-Barré syndrome when it reveals enhancement of the spinal nerve roots or cauda equina.
Other polyneuropathies
Polyneuropathy is caused by a variety of diseases that affect the function of peripheral motor, sensory, or autonomic nerves. The differential diagnosis is broad and involves inflammatory diseases (including autoimmune and paraneoplastic causes), hereditary disorders, infection, toxicity, and ischemic and nutritional deficiencies.5 Polyneuropathy can present in a distal-predominant, generalized, or asymmetric pattern involving individual nerve trunks termed “mononeuropathy multiplex,” as in our patient’s presentation. The initial workup includes EMG and a battery of serologic tests. In cases of severe and progressive polyneuropathy, nerve biopsy can assess for the presence of vasculitis, amyloidosis, and paraprotein deposition.
Transverse myelitis
Transverse myelitis is an inflammatory myelopathy that usually presents with acute or subacute weakness of the upper extremities or lower extremities, or both, corresponding to the level of the lesion, hyperreflexia, bladder and bowel dysfunction, spinal level of sensory loss, and autonomic involvement.6 The differential diagnosis of acute myelopathy includes:
- Infection (eg, herpes simplex virus, West Nile virus, Lyme disease, Mycoplasma pneumoniae, human immunodeficiency virus)
- Systemic inflammatory disease (systemic lupus erythematosus, sarcoidosis, Sjögren syndrome, scleroderma, paraneoplastic syndrome)
- Central nervous system demyelinating disease (acute disseminated encephalomyelitis, multiple sclerosis, neuromyelitis optica)
- Vascular malformation (dural arteriovenous fistula)
- Compression due to tumor, bleeding, disc herniation, infection, or abscess.
The workup involves laboratory tests to exclude systemic inflammatory and infectious causes, as well as MRI of the spine with and without contrast to identify a causative lesion. Lumbar puncture and CSF analysis may show pleocytosis, elevated protein concentration, and increased intrathecal immunoglobulin G (IgG) index.7
Although our patient’s presentation with subacute lower-extremity weakness, sensory changes, and bladder dysfunction were consistent with transverse myelitis, her cranial nerve abnormalities would be atypical for it.
Polyradiculopathy
Polyradiculopathy has many possible causes. In the United States, the most common causes are lumbar spondylosis, lumbar canal stenosis, and diabetic polyradiculoneuropathy.
When multiple spinal segments are affected, leptomeningeal disease involving the arachnoid and pia mater should be considered. Causes include malignant invasion, inflammatory cell accumulation, and protein deposition, leading to patchy but widespread dysfunction of spinal nerve roots and cranial nerves. Specific causes are myriad and include carcinomatous meningitis,8 syphilis, tuberculosis, sarcoidosis, and paraproteinemias. CSF and MRI changes are often nonspecific, leading to the need for meningeal biopsy for diagnosis.
CASE CONTINUED
During her hospitalization, our patient developed acute right upper and lower facial weakness consistent with peripheral facial mononeuropathy. Bilateral lower-extremity weakness progressed to disabling paraparesis.

She underwent lumbar puncture and CSF analysis (Table 1). The most notable findings were significant pleocytosis (72% lymphocytic predominance), protein elevation, and elevated IgG index (indicative of elevated intrathecal immunoglobulin synthesis in the central nervous system). Viral, bacterial, and fungal studies were negative. Guillain-Barré syndrome, other polyneuropathies, and spinal cord infarction would not be expected with these CSF features.
Surface EMG demonstrated normal sensory responses, and needle EMG showed chronic and active motor axon loss in the L3 and S1 root distributions, suggesting polyradiculopathy without polyneuropathy. These findings would not be expected in typical acute transverse myelitis but could be seen with spinal cord infarction.
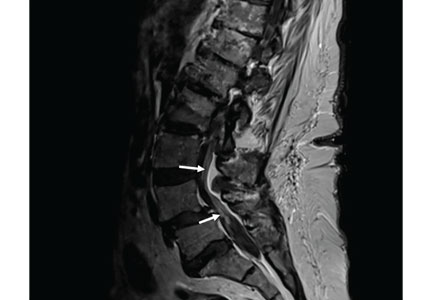
 in axial T1 sequence (top) and sagittal T1 sequence (bottom).")
MRI of the entire spine with and without contrast showed cauda equina nerve root thickening and enhancement, especially involving the L5 and S1 roots (Figure 1). The spinal cord appeared normal. These findings further supported polyradiculopathy and a leptomeningeal process.
Further evaluation included chest radiography, erythrocyte sedimentation rate, C-reactive protein, hemoglobin A1c, human immunodeficiency virus testing, antinuclear antibody, antineutrophil cytoplasmic antibody, extractable nuclear antibody, GQ1b antibody, serum and CSF paraneoplastic panels, levels of vitamin B1, B12, and B6, copper, and ceruloplasmin, and a screen for heavy metals. All results were within normal ranges.
ESTABLISHING THE DIAGNOSIS
Serum monoclonal protein analysis with immunofixation revealed IgM kappa monoclonal gammopathy with an IgM level of 1,570 (reference range 53–334 mg/dL) and M-spike 0.75 (0.00 mg/dL), serum free kappa light chains 61.1 (3.30–19.40 mg/L), lambda 9.3 (5.7–26.3 mg/L), and kappa-lambda ratio 6.57 (0.26–1.65).
2. Which is the best next step in this patient’s neurologic evaluation?
- Test CSF angiotensin-converting enzyme level
- CSF cytology
- Meningeal biopsy
- Peripheral nerve biopsy
Given the high suspicion for malignancy, CSF cytology was performed and showed increased numbers of mononuclear chronic inflammatory cells, including a mixture of lymphocytes and monocytes, favoring a reactive lymphoid pleocytosis. Flow cytometry indicated the presence of a monoclonal, CD5- and CD10- negative, B-cell lymphoproliferative disorder. The immunophenotypic findings were not specific for a single diagnosis. The differential diagnosis included marginal zone lymphoma and lymphoplasmacytic lymphoma.
3. Given the presence of serum IgM monoclonal gammopathy in this patient, which is the most likely diagnosis?
- Neurosarcoidosis
- Multiple myeloma
- Waldenström macroglobulinemia
- Carcinomatous meningitis
Study of bone marrow biopsy demonstrated limited bone marrow involvement (1%) by a lymphoproliferative disorder with plasmacytoid features, and DNA testing detected an MYD88 L265P mutation, reported to be present in 90% of patients with Waldenström macroglobulinemia.9 This finding confirmed the diagnosis of Waldenström macroglobulinemia with central nervous system involvement. Our patient began therapy with rituximab and methotrexate, which resulted in some improvement in strength, gait, and vision.
WALDENSTRÖM MACROGLOBULINEMIA AND BING-NEEL SYNDROME
Waldenström macroglobulinemia is a lymphoplasmacytic lymphoma associated with a monoclonal IgM protein.10 It is considered a paraproteinemic disorder, similar to multiple myeloma. The presenting symptoms and complications are related to direct tumor infiltration, hyperviscosity syndrome, and deposition of IgM in various tissues.11,12
Waldenström macroglobulinemia is usually indolent, and treatment is reserved for patients with symptoms.13,14 It includes rituximab, usually in combination with chemotherapy or other targeted agents.15,16
Paraneoplastic antibody-mediated polyneuropathy may occur in these patients. However, the pattern is usually symmetrical clinically, with demyelination on EMG, and is not associated with cranial nerve or meningeal involvement. Management with plasmapheresis, corticosteroids, and intravenous immunoglobulin has not been shown to be effective.17
Involvement of the central nervous system as a complication of Waldenström macroglobulinemia has been described as Bing-Neel syndrome. It can present as diffuse malignant cell infiltration of the leptomeningeal space, white matter, or spinal cord, or in a tumoral form presenting as intraparenchymal masses or nodular lesions. The distinction between the tumoral and diffuse forms is based primarily on imaging findings.18
In a report of 44 patients with Bing-Neel syndrome, 36% presented with the disorder as the initial manifestation of Waldenström macroglobulinemia.18 The primary presenting symptoms were imbalance and gait difficulty (48%) and cranial nerve involvement (36%), which presented as predominantly facial or oculomotor nerve palsy. Cauda equina syndrome with motor involvement (seen in our patient) occurred in 14% of patients. Other presenting symptoms included cognitive impairment, sensory deficits, headache, dysarthria, aphasia, and seizures.
LEARNING POINTS
The differential diagnosis for patients presenting with multifocal neurologic symptoms can be broad, and a systematic approach to the diagnosis is necessary. Localizing the lesion is important in determining the diagnosis for patients presenting with neurologic symptoms. The process of localization begins with taking the history, is further refined during the examination, and is confirmed with diagnostic studies. Atypical presentations of relatively common neurologic diseases such as Guillain-Barré syndrome, transverse myelitis, and peripheral polyneuropathy do occur, but uncommon diagnoses need to be considered when support for the initial diagnosis is lacking.
- Fokke C, van den Berg B, Drenthen J, Walgaard C, van Doorn PA, Jacobs BC. Diagnosis of Guillain-Barre syndrome and validation of Brighton criteria. Brain 2014; 137(Pt 1):33–43. doi:10.1093/brain/awt285
- Mathey EK, Park SB, Hughes RA, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry 2015; 86(9):973–985. doi:10.1136/jnnp-2014-309697
- Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barré syndrome: clinical and immunohistochemical studies. Neurology 1993; 43(10):1911–1917. pmid:8413947
- Teener J. Miller Fisher’s syndrome. Semin Neurol 2012; 32(5):512–516. doi:10.1055/s-0033-1334470
- Watson JC, Dyck PJ. Peripheral neuropathy: a practical approach to diagnosis and symptom management. Mayo Clin Proc 2015; 90(7):940–951. doi:10.1016/j.mayocp.2015.05.004
- Greenberg BM. Treatment of acute transverse myelitis and its early complications. Continuum (Minneap Minn) 2011; 17(4):733–743. doi:10.1212/01.CON.0000403792.36161.f5
- West TW. Transverse myelitis—a review of the presentation, diagnosis, and initial management. Discov Med 2013; 16(88):167–177. pmid:24099672
- Le Rhun E, Taillibert S, Chamberlain MC. Carcinomatous meningitis: leptomeningeal metastases in solid tumors. Surg Neurol Int 2013; 4(suppl 4):S265–S288. doi:10.4103/2152-7806.111304
- Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med 2012; 367(9):826–833. doi:10.1056/NEJMoa1200710
- Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol 2003; 30(2):110–115. doi:10.1053/sonc.2003.50082
- Björkholm M, Johansson E, Papamichael D, et al. Patterns of clinical presentation, treatment, and outcome in patients with Waldenstrom’s macroglobulinemia: a two-institution study. Semin Oncol 2003; 30(2):226–230. doi:10.1053/sonc.2003.50054
- Rison RA, Beydoun SR. Paraproteinemic neuropathy: a practical review. BMC Neurol 2016; 16:13. doi:10.1186/s12883-016-0532-4
- Kyle RA, Benson J, Larson D, et al. IgM monoclonal gammopathy of undetermined significance and smoldering Waldenström’s macroglobulinemia. Clin Lymphoma Myeloma 2009; 9(1):17–18. doi:10.3816/CLM.2009.n.002
- Kyle RA, Benson JT, Larson DR, et al. Progression in smoldering Waldenstrom macroglobulinemia: long-term results. Blood 2012; 119(19):4462–4466. doi:10.1182/blood-2011-10-384768
- Leblond V, Kastritis E, Advani R, et al. Treatment recommendations from the Eighth International Workshop on Waldenström’s macroglobulinemia. Blood 2016; 128(10):1321–1328. doi:10.1182/blood-2016-04-711234
- Kapoor P, Ansell SM, Fonseca R, et al. Diagnosis and management of Waldenström macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines 2016. JAMA Oncol 2017; 3(9):1257–1265. doi:10.1001/jamaoncol.2016.5763
- D’Sa S, Kersten MJ, Castillo JJ, et al. Investigation and management of IgM and Waldenström-associated peripheral neuropathies: recommendations from the IWWM-8 consensus panel. Br J Haematol 2017; 176(5):728–742. doi:10.1111/bjh.14492
- Simon L, Fitsiori A, Lemal R, et al. Bing-Neel syndrome, a rare complication of Waldenström macroglobulinemia: analysis of 44 cases and review of the literature. A study on behalf of the French Innovative Leukemia Organization (FILO). Haematologica 2015; 100(12):1587–1594. doi:10.3324/haematol.2015.133744
A 69-year-old woman was admitted to the hospital with double vision, weakness in the lower extremities, sensory loss, pain, and falls. Her symptoms started with sudden onset of horizontal diplopia 6 weeks before, followed by gradually worsening lower-extremity weakness, as well as ataxia and patchy and bilateral radicular burning leg pain more pronounced on the right. Her medical history included narcolepsy, obstructive sleep apnea, hypertension, hyperlipidemia, and bilateral knee replacements for osteoarthritis.
Neurologic examination showed inability to abduct the right eye, bilateral hip flexion weakness, decreased pinprick response, decreased proprioception, and diminished muscle stretch reflexes in the lower extremities. Magnetic resonance imaging (MRI) of the brain without contrast and magnetic resonance angiography of the brain and carotid arteries showed no evidence of acute stroke. No abnormalities were noted on electrocardiography and echocardiography.
A diagnosis of idiopathic peripheral neuropathy was made, and outpatient physical therapy was recommended. Over the subsequent 2 weeks, her condition declined to the point where she needed a walker. She continued to have worsening leg weakness with falls, prompting hospital readmission.
INITIAL EVALUATION
In addition to her diplopia and weakness, she said she had lost 15 pounds since the onset of symptoms and had experienced symptoms suggesting urinary retention.
Physical examination
Her temperature was 37°C (98.6°F), heart rate 79 beats per minute, blood pressure 117/86 mm Hg, respiratory rate 14 breaths per minute, and oxygen saturation 98% on room air. Examination of the head, neck, heart, lung, abdomen, lymph nodes, and extremities yielded nothing remarkable except for chronic venous changes in the lower extremities.
The neurologic examination showed incomplete lateral gaze bilaterally (cranial nerve VI dysfunction). Strength in the upper extremities was normal. In the legs, the Medical Research Council scale score for proximal muscle strength was 2 to 3 out of 5, and for distal muscles 3 to 4 out of 5, with the right side worse than the left and flexors and extensors affected equally. Muscle stretch reflexes were absent in both lower extremities and the left upper extremity, but intact in the right upper extremity. No abnormal corticospinal tract reflexes were elicited.
Sensory testing revealed diminished pin-prick perception in a length-dependent fashion in the lower extremities, reduced 50% compared with the hands. Gait could not be assessed due to weakness.
Initial laboratory testing
Results of initial laboratory tests—complete blood cell count, complete metabolic panel, erythrocyte sedimentation rate, C-reactive protein, thyroid-stimulating hormone, and hemoglobin A1c—were unremarkable.
FURTHER EVALUATION AND DIFFERENTIAL DIAGNOSIS
1. Which of the following is the most likely diagnosis at this point?
- Cerebral infarction
- Guillain-Barré syndrome
- Progressive polyneuropathy
- Transverse myelitis
- Polyradiculopathy
In the absence of definitive diagnostic tests, all of the above options were considered in the differential diagnosis for this patient.
Cerebral infarction
Although acute-onset diplopia can be explained by brainstem stroke involving cranial nerve nuclei or their projections, the onset of diplopia with progressive bilateral lower-extremity weakness makes stroke unlikely. Flaccid paralysis, areflexia of the lower extremities, and sensory involvement can also be caused by acute anterior spinal artery occlusion leading to spinal cord infarction; however, the deficits are usually maximal at onset.
Guillain-Barré syndrome
The combination of acute-subacute progressive ascending weakness, sensory involvement, and diminished or absent reflexes is typical of Guillain-Barré syndrome. Cranial nerve involvement can overlap with the more typical features of the syndrome. However, most patients reach the nadir of their disease by 4 weeks after initial symptom onset, even without treatment.1 This patient’s condition continued to worsen over 8 weeks. In addition, the asymmetric lower-extremity weakness and sparing of the arms are atypical for Guillain-Barré syndrome.
Given the progression of symptoms, chronic inflammatory demyelinating polyneuropathy is also a consideration, typically presenting as a relapsing or progressive neuropathy in proximal and distal muscles and worsening over at least an 8-week period.2
The initial workup for Guillain-Barré syndrome or chronic inflammatory demyelinating polyneuropathy includes lumbar puncture to assess for albuminocytologic dissociation (elevated protein with normal white blood cell count) in cerebrospinal fluid (CSF), and electromyography (EMG) to assess for neurophysiologic evidence of peripheral nerve demyelination. In Miller-Fisher syndrome, a rare variant of Guillain-Barré syndrome characterized by ataxia, ophthalmoparesis, and areflexia, serum ganglioside antibodies to GQ1b are found in over 90% of patients.3,4 Although MRI of the spine is not necessary to diagnose Guillain-Barré syndrome, it is often done to exclude other causes of lower-extremity weakness such as spinal cord or cauda equina compression that would require urgent neurosurgical consultation. MRI can support the diagnosis of Guillain-Barré syndrome when it reveals enhancement of the spinal nerve roots or cauda equina.
Other polyneuropathies
Polyneuropathy is caused by a variety of diseases that affect the function of peripheral motor, sensory, or autonomic nerves. The differential diagnosis is broad and involves inflammatory diseases (including autoimmune and paraneoplastic causes), hereditary disorders, infection, toxicity, and ischemic and nutritional deficiencies.5 Polyneuropathy can present in a distal-predominant, generalized, or asymmetric pattern involving individual nerve trunks termed “mononeuropathy multiplex,” as in our patient’s presentation. The initial workup includes EMG and a battery of serologic tests. In cases of severe and progressive polyneuropathy, nerve biopsy can assess for the presence of vasculitis, amyloidosis, and paraprotein deposition.
Transverse myelitis
Transverse myelitis is an inflammatory myelopathy that usually presents with acute or subacute weakness of the upper extremities or lower extremities, or both, corresponding to the level of the lesion, hyperreflexia, bladder and bowel dysfunction, spinal level of sensory loss, and autonomic involvement.6 The differential diagnosis of acute myelopathy includes:
- Infection (eg, herpes simplex virus, West Nile virus, Lyme disease, Mycoplasma pneumoniae, human immunodeficiency virus)
- Systemic inflammatory disease (systemic lupus erythematosus, sarcoidosis, Sjögren syndrome, scleroderma, paraneoplastic syndrome)
- Central nervous system demyelinating disease (acute disseminated encephalomyelitis, multiple sclerosis, neuromyelitis optica)
- Vascular malformation (dural arteriovenous fistula)
- Compression due to tumor, bleeding, disc herniation, infection, or abscess.
The workup involves laboratory tests to exclude systemic inflammatory and infectious causes, as well as MRI of the spine with and without contrast to identify a causative lesion. Lumbar puncture and CSF analysis may show pleocytosis, elevated protein concentration, and increased intrathecal immunoglobulin G (IgG) index.7
Although our patient’s presentation with subacute lower-extremity weakness, sensory changes, and bladder dysfunction were consistent with transverse myelitis, her cranial nerve abnormalities would be atypical for it.
Polyradiculopathy
Polyradiculopathy has many possible causes. In the United States, the most common causes are lumbar spondylosis, lumbar canal stenosis, and diabetic polyradiculoneuropathy.
When multiple spinal segments are affected, leptomeningeal disease involving the arachnoid and pia mater should be considered. Causes include malignant invasion, inflammatory cell accumulation, and protein deposition, leading to patchy but widespread dysfunction of spinal nerve roots and cranial nerves. Specific causes are myriad and include carcinomatous meningitis,8 syphilis, tuberculosis, sarcoidosis, and paraproteinemias. CSF and MRI changes are often nonspecific, leading to the need for meningeal biopsy for diagnosis.
CASE CONTINUED
During her hospitalization, our patient developed acute right upper and lower facial weakness consistent with peripheral facial mononeuropathy. Bilateral lower-extremity weakness progressed to disabling paraparesis.
She underwent lumbar puncture and CSF analysis (Table 1). The most notable findings were significant pleocytosis (72% lymphocytic predominance), protein elevation, and elevated IgG index (indicative of elevated intrathecal immunoglobulin synthesis in the central nervous system). Viral, bacterial, and fungal studies were negative. Guillain-Barré syndrome, other polyneuropathies, and spinal cord infarction would not be expected with these CSF features.
Surface EMG demonstrated normal sensory responses, and needle EMG showed chronic and active motor axon loss in the L3 and S1 root distributions, suggesting polyradiculopathy without polyneuropathy. These findings would not be expected in typical acute transverse myelitis but could be seen with spinal cord infarction.
MRI of the entire spine with and without contrast showed cauda equina nerve root thickening and enhancement, especially involving the L5 and S1 roots (Figure 1). The spinal cord appeared normal. These findings further supported polyradiculopathy and a leptomeningeal process.
Further evaluation included chest radiography, erythrocyte sedimentation rate, C-reactive protein, hemoglobin A1c, human immunodeficiency virus testing, antinuclear antibody, antineutrophil cytoplasmic antibody, extractable nuclear antibody, GQ1b antibody, serum and CSF paraneoplastic panels, levels of vitamin B1, B12, and B6, copper, and ceruloplasmin, and a screen for heavy metals. All results were within normal ranges.
ESTABLISHING THE DIAGNOSIS
Serum monoclonal protein analysis with immunofixation revealed IgM kappa monoclonal gammopathy with an IgM level of 1,570 (reference range 53–334 mg/dL) and M-spike 0.75 (0.00 mg/dL), serum free kappa light chains 61.1 (3.30–19.40 mg/L), lambda 9.3 (5.7–26.3 mg/L), and kappa-lambda ratio 6.57 (0.26–1.65).
2. Which is the best next step in this patient’s neurologic evaluation?
- Test CSF angiotensin-converting enzyme level
- CSF cytology
- Meningeal biopsy
- Peripheral nerve biopsy
Given the high suspicion for malignancy, CSF cytology was performed and showed increased numbers of mononuclear chronic inflammatory cells, including a mixture of lymphocytes and monocytes, favoring a reactive lymphoid pleocytosis. Flow cytometry indicated the presence of a monoclonal, CD5- and CD10- negative, B-cell lymphoproliferative disorder. The immunophenotypic findings were not specific for a single diagnosis. The differential diagnosis included marginal zone lymphoma and lymphoplasmacytic lymphoma.
3. Given the presence of serum IgM monoclonal gammopathy in this patient, which is the most likely diagnosis?
- Neurosarcoidosis
- Multiple myeloma
- Waldenström macroglobulinemia
- Carcinomatous meningitis
Study of bone marrow biopsy demonstrated limited bone marrow involvement (1%) by a lymphoproliferative disorder with plasmacytoid features, and DNA testing detected an MYD88 L265P mutation, reported to be present in 90% of patients with Waldenström macroglobulinemia.9 This finding confirmed the diagnosis of Waldenström macroglobulinemia with central nervous system involvement. Our patient began therapy with rituximab and methotrexate, which resulted in some improvement in strength, gait, and vision.
WALDENSTRÖM MACROGLOBULINEMIA AND BING-NEEL SYNDROME
Waldenström macroglobulinemia is a lymphoplasmacytic lymphoma associated with a monoclonal IgM protein.10 It is considered a paraproteinemic disorder, similar to multiple myeloma. The presenting symptoms and complications are related to direct tumor infiltration, hyperviscosity syndrome, and deposition of IgM in various tissues.11,12
Waldenström macroglobulinemia is usually indolent, and treatment is reserved for patients with symptoms.13,14 It includes rituximab, usually in combination with chemotherapy or other targeted agents.15,16
Paraneoplastic antibody-mediated polyneuropathy may occur in these patients. However, the pattern is usually symmetrical clinically, with demyelination on EMG, and is not associated with cranial nerve or meningeal involvement. Management with plasmapheresis, corticosteroids, and intravenous immunoglobulin has not been shown to be effective.17
Involvement of the central nervous system as a complication of Waldenström macroglobulinemia has been described as Bing-Neel syndrome. It can present as diffuse malignant cell infiltration of the leptomeningeal space, white matter, or spinal cord, or in a tumoral form presenting as intraparenchymal masses or nodular lesions. The distinction between the tumoral and diffuse forms is based primarily on imaging findings.18
In a report of 44 patients with Bing-Neel syndrome, 36% presented with the disorder as the initial manifestation of Waldenström macroglobulinemia.18 The primary presenting symptoms were imbalance and gait difficulty (48%) and cranial nerve involvement (36%), which presented as predominantly facial or oculomotor nerve palsy. Cauda equina syndrome with motor involvement (seen in our patient) occurred in 14% of patients. Other presenting symptoms included cognitive impairment, sensory deficits, headache, dysarthria, aphasia, and seizures.
LEARNING POINTS
The differential diagnosis for patients presenting with multifocal neurologic symptoms can be broad, and a systematic approach to the diagnosis is necessary. Localizing the lesion is important in determining the diagnosis for patients presenting with neurologic symptoms. The process of localization begins with taking the history, is further refined during the examination, and is confirmed with diagnostic studies. Atypical presentations of relatively common neurologic diseases such as Guillain-Barré syndrome, transverse myelitis, and peripheral polyneuropathy do occur, but uncommon diagnoses need to be considered when support for the initial diagnosis is lacking.
A 69-year-old woman was admitted to the hospital with double vision, weakness in the lower extremities, sensory loss, pain, and falls. Her symptoms started with sudden onset of horizontal diplopia 6 weeks before, followed by gradually worsening lower-extremity weakness, as well as ataxia and patchy and bilateral radicular burning leg pain more pronounced on the right. Her medical history included narcolepsy, obstructive sleep apnea, hypertension, hyperlipidemia, and bilateral knee replacements for osteoarthritis.
Neurologic examination showed inability to abduct the right eye, bilateral hip flexion weakness, decreased pinprick response, decreased proprioception, and diminished muscle stretch reflexes in the lower extremities. Magnetic resonance imaging (MRI) of the brain without contrast and magnetic resonance angiography of the brain and carotid arteries showed no evidence of acute stroke. No abnormalities were noted on electrocardiography and echocardiography.
A diagnosis of idiopathic peripheral neuropathy was made, and outpatient physical therapy was recommended. Over the subsequent 2 weeks, her condition declined to the point where she needed a walker. She continued to have worsening leg weakness with falls, prompting hospital readmission.
INITIAL EVALUATION
In addition to her diplopia and weakness, she said she had lost 15 pounds since the onset of symptoms and had experienced symptoms suggesting urinary retention.
Physical examination
Her temperature was 37°C (98.6°F), heart rate 79 beats per minute, blood pressure 117/86 mm Hg, respiratory rate 14 breaths per minute, and oxygen saturation 98% on room air. Examination of the head, neck, heart, lung, abdomen, lymph nodes, and extremities yielded nothing remarkable except for chronic venous changes in the lower extremities.
The neurologic examination showed incomplete lateral gaze bilaterally (cranial nerve VI dysfunction). Strength in the upper extremities was normal. In the legs, the Medical Research Council scale score for proximal muscle strength was 2 to 3 out of 5, and for distal muscles 3 to 4 out of 5, with the right side worse than the left and flexors and extensors affected equally. Muscle stretch reflexes were absent in both lower extremities and the left upper extremity, but intact in the right upper extremity. No abnormal corticospinal tract reflexes were elicited.
Sensory testing revealed diminished pin-prick perception in a length-dependent fashion in the lower extremities, reduced 50% compared with the hands. Gait could not be assessed due to weakness.
Initial laboratory testing
Results of initial laboratory tests—complete blood cell count, complete metabolic panel, erythrocyte sedimentation rate, C-reactive protein, thyroid-stimulating hormone, and hemoglobin A1c—were unremarkable.
FURTHER EVALUATION AND DIFFERENTIAL DIAGNOSIS
1. Which of the following is the most likely diagnosis at this point?
- Cerebral infarction
- Guillain-Barré syndrome
- Progressive polyneuropathy
- Transverse myelitis
- Polyradiculopathy
In the absence of definitive diagnostic tests, all of the above options were considered in the differential diagnosis for this patient.
Cerebral infarction
Although acute-onset diplopia can be explained by brainstem stroke involving cranial nerve nuclei or their projections, the onset of diplopia with progressive bilateral lower-extremity weakness makes stroke unlikely. Flaccid paralysis, areflexia of the lower extremities, and sensory involvement can also be caused by acute anterior spinal artery occlusion leading to spinal cord infarction; however, the deficits are usually maximal at onset.
Guillain-Barré syndrome
The combination of acute-subacute progressive ascending weakness, sensory involvement, and diminished or absent reflexes is typical of Guillain-Barré syndrome. Cranial nerve involvement can overlap with the more typical features of the syndrome. However, most patients reach the nadir of their disease by 4 weeks after initial symptom onset, even without treatment.1 This patient’s condition continued to worsen over 8 weeks. In addition, the asymmetric lower-extremity weakness and sparing of the arms are atypical for Guillain-Barré syndrome.
Given the progression of symptoms, chronic inflammatory demyelinating polyneuropathy is also a consideration, typically presenting as a relapsing or progressive neuropathy in proximal and distal muscles and worsening over at least an 8-week period.2
The initial workup for Guillain-Barré syndrome or chronic inflammatory demyelinating polyneuropathy includes lumbar puncture to assess for albuminocytologic dissociation (elevated protein with normal white blood cell count) in cerebrospinal fluid (CSF), and electromyography (EMG) to assess for neurophysiologic evidence of peripheral nerve demyelination. In Miller-Fisher syndrome, a rare variant of Guillain-Barré syndrome characterized by ataxia, ophthalmoparesis, and areflexia, serum ganglioside antibodies to GQ1b are found in over 90% of patients.3,4 Although MRI of the spine is not necessary to diagnose Guillain-Barré syndrome, it is often done to exclude other causes of lower-extremity weakness such as spinal cord or cauda equina compression that would require urgent neurosurgical consultation. MRI can support the diagnosis of Guillain-Barré syndrome when it reveals enhancement of the spinal nerve roots or cauda equina.
Other polyneuropathies
Polyneuropathy is caused by a variety of diseases that affect the function of peripheral motor, sensory, or autonomic nerves. The differential diagnosis is broad and involves inflammatory diseases (including autoimmune and paraneoplastic causes), hereditary disorders, infection, toxicity, and ischemic and nutritional deficiencies.5 Polyneuropathy can present in a distal-predominant, generalized, or asymmetric pattern involving individual nerve trunks termed “mononeuropathy multiplex,” as in our patient’s presentation. The initial workup includes EMG and a battery of serologic tests. In cases of severe and progressive polyneuropathy, nerve biopsy can assess for the presence of vasculitis, amyloidosis, and paraprotein deposition.
Transverse myelitis
Transverse myelitis is an inflammatory myelopathy that usually presents with acute or subacute weakness of the upper extremities or lower extremities, or both, corresponding to the level of the lesion, hyperreflexia, bladder and bowel dysfunction, spinal level of sensory loss, and autonomic involvement.6 The differential diagnosis of acute myelopathy includes:
- Infection (eg, herpes simplex virus, West Nile virus, Lyme disease, Mycoplasma pneumoniae, human immunodeficiency virus)
- Systemic inflammatory disease (systemic lupus erythematosus, sarcoidosis, Sjögren syndrome, scleroderma, paraneoplastic syndrome)
- Central nervous system demyelinating disease (acute disseminated encephalomyelitis, multiple sclerosis, neuromyelitis optica)
- Vascular malformation (dural arteriovenous fistula)
- Compression due to tumor, bleeding, disc herniation, infection, or abscess.
The workup involves laboratory tests to exclude systemic inflammatory and infectious causes, as well as MRI of the spine with and without contrast to identify a causative lesion. Lumbar puncture and CSF analysis may show pleocytosis, elevated protein concentration, and increased intrathecal immunoglobulin G (IgG) index.7
Although our patient’s presentation with subacute lower-extremity weakness, sensory changes, and bladder dysfunction were consistent with transverse myelitis, her cranial nerve abnormalities would be atypical for it.
Polyradiculopathy
Polyradiculopathy has many possible causes. In the United States, the most common causes are lumbar spondylosis, lumbar canal stenosis, and diabetic polyradiculoneuropathy.
When multiple spinal segments are affected, leptomeningeal disease involving the arachnoid and pia mater should be considered. Causes include malignant invasion, inflammatory cell accumulation, and protein deposition, leading to patchy but widespread dysfunction of spinal nerve roots and cranial nerves. Specific causes are myriad and include carcinomatous meningitis,8 syphilis, tuberculosis, sarcoidosis, and paraproteinemias. CSF and MRI changes are often nonspecific, leading to the need for meningeal biopsy for diagnosis.
CASE CONTINUED
During her hospitalization, our patient developed acute right upper and lower facial weakness consistent with peripheral facial mononeuropathy. Bilateral lower-extremity weakness progressed to disabling paraparesis.
She underwent lumbar puncture and CSF analysis (Table 1). The most notable findings were significant pleocytosis (72% lymphocytic predominance), protein elevation, and elevated IgG index (indicative of elevated intrathecal immunoglobulin synthesis in the central nervous system). Viral, bacterial, and fungal studies were negative. Guillain-Barré syndrome, other polyneuropathies, and spinal cord infarction would not be expected with these CSF features.
Surface EMG demonstrated normal sensory responses, and needle EMG showed chronic and active motor axon loss in the L3 and S1 root distributions, suggesting polyradiculopathy without polyneuropathy. These findings would not be expected in typical acute transverse myelitis but could be seen with spinal cord infarction.
MRI of the entire spine with and without contrast showed cauda equina nerve root thickening and enhancement, especially involving the L5 and S1 roots (Figure 1). The spinal cord appeared normal. These findings further supported polyradiculopathy and a leptomeningeal process.
Further evaluation included chest radiography, erythrocyte sedimentation rate, C-reactive protein, hemoglobin A1c, human immunodeficiency virus testing, antinuclear antibody, antineutrophil cytoplasmic antibody, extractable nuclear antibody, GQ1b antibody, serum and CSF paraneoplastic panels, levels of vitamin B1, B12, and B6, copper, and ceruloplasmin, and a screen for heavy metals. All results were within normal ranges.
ESTABLISHING THE DIAGNOSIS
Serum monoclonal protein analysis with immunofixation revealed IgM kappa monoclonal gammopathy with an IgM level of 1,570 (reference range 53–334 mg/dL) and M-spike 0.75 (0.00 mg/dL), serum free kappa light chains 61.1 (3.30–19.40 mg/L), lambda 9.3 (5.7–26.3 mg/L), and kappa-lambda ratio 6.57 (0.26–1.65).
2. Which is the best next step in this patient’s neurologic evaluation?
- Test CSF angiotensin-converting enzyme level
- CSF cytology
- Meningeal biopsy
- Peripheral nerve biopsy
Given the high suspicion for malignancy, CSF cytology was performed and showed increased numbers of mononuclear chronic inflammatory cells, including a mixture of lymphocytes and monocytes, favoring a reactive lymphoid pleocytosis. Flow cytometry indicated the presence of a monoclonal, CD5- and CD10- negative, B-cell lymphoproliferative disorder. The immunophenotypic findings were not specific for a single diagnosis. The differential diagnosis included marginal zone lymphoma and lymphoplasmacytic lymphoma.
3. Given the presence of serum IgM monoclonal gammopathy in this patient, which is the most likely diagnosis?
- Neurosarcoidosis
- Multiple myeloma
- Waldenström macroglobulinemia
- Carcinomatous meningitis
Study of bone marrow biopsy demonstrated limited bone marrow involvement (1%) by a lymphoproliferative disorder with plasmacytoid features, and DNA testing detected an MYD88 L265P mutation, reported to be present in 90% of patients with Waldenström macroglobulinemia.9 This finding confirmed the diagnosis of Waldenström macroglobulinemia with central nervous system involvement. Our patient began therapy with rituximab and methotrexate, which resulted in some improvement in strength, gait, and vision.
WALDENSTRÖM MACROGLOBULINEMIA AND BING-NEEL SYNDROME
Waldenström macroglobulinemia is a lymphoplasmacytic lymphoma associated with a monoclonal IgM protein.10 It is considered a paraproteinemic disorder, similar to multiple myeloma. The presenting symptoms and complications are related to direct tumor infiltration, hyperviscosity syndrome, and deposition of IgM in various tissues.11,12
Waldenström macroglobulinemia is usually indolent, and treatment is reserved for patients with symptoms.13,14 It includes rituximab, usually in combination with chemotherapy or other targeted agents.15,16
Paraneoplastic antibody-mediated polyneuropathy may occur in these patients. However, the pattern is usually symmetrical clinically, with demyelination on EMG, and is not associated with cranial nerve or meningeal involvement. Management with plasmapheresis, corticosteroids, and intravenous immunoglobulin has not been shown to be effective.17
Involvement of the central nervous system as a complication of Waldenström macroglobulinemia has been described as Bing-Neel syndrome. It can present as diffuse malignant cell infiltration of the leptomeningeal space, white matter, or spinal cord, or in a tumoral form presenting as intraparenchymal masses or nodular lesions. The distinction between the tumoral and diffuse forms is based primarily on imaging findings.18
In a report of 44 patients with Bing-Neel syndrome, 36% presented with the disorder as the initial manifestation of Waldenström macroglobulinemia.18 The primary presenting symptoms were imbalance and gait difficulty (48%) and cranial nerve involvement (36%), which presented as predominantly facial or oculomotor nerve palsy. Cauda equina syndrome with motor involvement (seen in our patient) occurred in 14% of patients. Other presenting symptoms included cognitive impairment, sensory deficits, headache, dysarthria, aphasia, and seizures.
LEARNING POINTS
The differential diagnosis for patients presenting with multifocal neurologic symptoms can be broad, and a systematic approach to the diagnosis is necessary. Localizing the lesion is important in determining the diagnosis for patients presenting with neurologic symptoms. The process of localization begins with taking the history, is further refined during the examination, and is confirmed with diagnostic studies. Atypical presentations of relatively common neurologic diseases such as Guillain-Barré syndrome, transverse myelitis, and peripheral polyneuropathy do occur, but uncommon diagnoses need to be considered when support for the initial diagnosis is lacking.
- Fokke C, van den Berg B, Drenthen J, Walgaard C, van Doorn PA, Jacobs BC. Diagnosis of Guillain-Barre syndrome and validation of Brighton criteria. Brain 2014; 137(Pt 1):33–43. doi:10.1093/brain/awt285
- Mathey EK, Park SB, Hughes RA, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry 2015; 86(9):973–985. doi:10.1136/jnnp-2014-309697
- Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barré syndrome: clinical and immunohistochemical studies. Neurology 1993; 43(10):1911–1917. pmid:8413947
- Teener J. Miller Fisher’s syndrome. Semin Neurol 2012; 32(5):512–516. doi:10.1055/s-0033-1334470
- Watson JC, Dyck PJ. Peripheral neuropathy: a practical approach to diagnosis and symptom management. Mayo Clin Proc 2015; 90(7):940–951. doi:10.1016/j.mayocp.2015.05.004
- Greenberg BM. Treatment of acute transverse myelitis and its early complications. Continuum (Minneap Minn) 2011; 17(4):733–743. doi:10.1212/01.CON.0000403792.36161.f5
- West TW. Transverse myelitis—a review of the presentation, diagnosis, and initial management. Discov Med 2013; 16(88):167–177. pmid:24099672
- Le Rhun E, Taillibert S, Chamberlain MC. Carcinomatous meningitis: leptomeningeal metastases in solid tumors. Surg Neurol Int 2013; 4(suppl 4):S265–S288. doi:10.4103/2152-7806.111304
- Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med 2012; 367(9):826–833. doi:10.1056/NEJMoa1200710
- Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol 2003; 30(2):110–115. doi:10.1053/sonc.2003.50082
- Björkholm M, Johansson E, Papamichael D, et al. Patterns of clinical presentation, treatment, and outcome in patients with Waldenstrom’s macroglobulinemia: a two-institution study. Semin Oncol 2003; 30(2):226–230. doi:10.1053/sonc.2003.50054
- Rison RA, Beydoun SR. Paraproteinemic neuropathy: a practical review. BMC Neurol 2016; 16:13. doi:10.1186/s12883-016-0532-4
- Kyle RA, Benson J, Larson D, et al. IgM monoclonal gammopathy of undetermined significance and smoldering Waldenström’s macroglobulinemia. Clin Lymphoma Myeloma 2009; 9(1):17–18. doi:10.3816/CLM.2009.n.002
- Kyle RA, Benson JT, Larson DR, et al. Progression in smoldering Waldenstrom macroglobulinemia: long-term results. Blood 2012; 119(19):4462–4466. doi:10.1182/blood-2011-10-384768
- Leblond V, Kastritis E, Advani R, et al. Treatment recommendations from the Eighth International Workshop on Waldenström’s macroglobulinemia. Blood 2016; 128(10):1321–1328. doi:10.1182/blood-2016-04-711234
- Kapoor P, Ansell SM, Fonseca R, et al. Diagnosis and management of Waldenström macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines 2016. JAMA Oncol 2017; 3(9):1257–1265. doi:10.1001/jamaoncol.2016.5763
- D’Sa S, Kersten MJ, Castillo JJ, et al. Investigation and management of IgM and Waldenström-associated peripheral neuropathies: recommendations from the IWWM-8 consensus panel. Br J Haematol 2017; 176(5):728–742. doi:10.1111/bjh.14492
- Simon L, Fitsiori A, Lemal R, et al. Bing-Neel syndrome, a rare complication of Waldenström macroglobulinemia: analysis of 44 cases and review of the literature. A study on behalf of the French Innovative Leukemia Organization (FILO). Haematologica 2015; 100(12):1587–1594. doi:10.3324/haematol.2015.133744
- Fokke C, van den Berg B, Drenthen J, Walgaard C, van Doorn PA, Jacobs BC. Diagnosis of Guillain-Barre syndrome and validation of Brighton criteria. Brain 2014; 137(Pt 1):33–43. doi:10.1093/brain/awt285
- Mathey EK, Park SB, Hughes RA, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry 2015; 86(9):973–985. doi:10.1136/jnnp-2014-309697
- Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barré syndrome: clinical and immunohistochemical studies. Neurology 1993; 43(10):1911–1917. pmid:8413947
- Teener J. Miller Fisher’s syndrome. Semin Neurol 2012; 32(5):512–516. doi:10.1055/s-0033-1334470
- Watson JC, Dyck PJ. Peripheral neuropathy: a practical approach to diagnosis and symptom management. Mayo Clin Proc 2015; 90(7):940–951. doi:10.1016/j.mayocp.2015.05.004
- Greenberg BM. Treatment of acute transverse myelitis and its early complications. Continuum (Minneap Minn) 2011; 17(4):733–743. doi:10.1212/01.CON.0000403792.36161.f5
- West TW. Transverse myelitis—a review of the presentation, diagnosis, and initial management. Discov Med 2013; 16(88):167–177. pmid:24099672
- Le Rhun E, Taillibert S, Chamberlain MC. Carcinomatous meningitis: leptomeningeal metastases in solid tumors. Surg Neurol Int 2013; 4(suppl 4):S265–S288. doi:10.4103/2152-7806.111304
- Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med 2012; 367(9):826–833. doi:10.1056/NEJMoa1200710
- Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol 2003; 30(2):110–115. doi:10.1053/sonc.2003.50082
- Björkholm M, Johansson E, Papamichael D, et al. Patterns of clinical presentation, treatment, and outcome in patients with Waldenstrom’s macroglobulinemia: a two-institution study. Semin Oncol 2003; 30(2):226–230. doi:10.1053/sonc.2003.50054
- Rison RA, Beydoun SR. Paraproteinemic neuropathy: a practical review. BMC Neurol 2016; 16:13. doi:10.1186/s12883-016-0532-4
- Kyle RA, Benson J, Larson D, et al. IgM monoclonal gammopathy of undetermined significance and smoldering Waldenström’s macroglobulinemia. Clin Lymphoma Myeloma 2009; 9(1):17–18. doi:10.3816/CLM.2009.n.002
- Kyle RA, Benson JT, Larson DR, et al. Progression in smoldering Waldenstrom macroglobulinemia: long-term results. Blood 2012; 119(19):4462–4466. doi:10.1182/blood-2011-10-384768
- Leblond V, Kastritis E, Advani R, et al. Treatment recommendations from the Eighth International Workshop on Waldenström’s macroglobulinemia. Blood 2016; 128(10):1321–1328. doi:10.1182/blood-2016-04-711234
- Kapoor P, Ansell SM, Fonseca R, et al. Diagnosis and management of Waldenström macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines 2016. JAMA Oncol 2017; 3(9):1257–1265. doi:10.1001/jamaoncol.2016.5763
- D’Sa S, Kersten MJ, Castillo JJ, et al. Investigation and management of IgM and Waldenström-associated peripheral neuropathies: recommendations from the IWWM-8 consensus panel. Br J Haematol 2017; 176(5):728–742. doi:10.1111/bjh.14492
- Simon L, Fitsiori A, Lemal R, et al. Bing-Neel syndrome, a rare complication of Waldenström macroglobulinemia: analysis of 44 cases and review of the literature. A study on behalf of the French Innovative Leukemia Organization (FILO). Haematologica 2015; 100(12):1587–1594. doi:10.3324/haematol.2015.133744