User login
Diagnosis and management of gastric intestinal metaplasia in the United States
Introduction
Despite a global decline in the incidence of gastric cancer over the past 3 decades, it remains the fifth most commonly diagnosed cancer and the third most common cause of cancer deaths worldwide.1 In the United States it is the fourth most commonly diagnosed GI malignancy, after colorectal, pancreas, and liver cancer. The prevalence remains high in Latin America and Asia, which has implications in the United States because of growing Hispanic and Asian populations.2,3 In recent years, a change in the trend of gastric cancer among non-Hispanic whites has been observed, particularly in women younger than 50 years old.4 5
Etiology
Gastric adenocarcinomas are classified into two subcategories based on location (cardia and noncardia) and histology (intestinal and diffuse types).6,7 Atrophic gastritis and gastric intestinal metaplasia (GIM) are considered precursors of intestinal-type noncardia gastric adenocarcinoma. The Correa cascade is a commonly accepted precancer sequence for noncardia gastric adenocarcinoma that describes mucosal changes from inflammation to atrophy to metaplasia to intraepithelial neoplasia and culminating in carcinoma.8,9 It has been observed that GIM may be the histologic change prior to the development of dysplasia and over 50% of patients with high-grade dysplasia will progress to adenocarcinoma.10-12 In the United States, GIM has the highest prevalence in African Americans, Hispanics, and East Asians, with the overall GIM prevalence regardless of ethnicity reported from 3.05% to 19.2%.5,13
Risk factors and subclassification
Replacement of the foveolar and/or glandular epithelium in the oxyntic and antral mucosa by intestinal epithelium results in GIM. It can be focal when limited to one region of the stomach or extensive when two or more regions are involved.14 The main risk factors for GIM development are Helicobacter pylori infection, tobacco, alcohol consumption, high salt intake, and chronic bile reflux.15,16 Additional risks for developing gastric cancer include older age, certain ethnicities, and male sex.17

CagA strains of H. pylori can promote carcinogenesis by inducing a mitogenic cellular response and downregulating cell adhesion.18,19 Less carcinogenic risk is associated with H. pylori Cag-A negative strains; however, they also have oncogenic potential mediated by expression of babA2 and vacA genes.20 Hence, the combination of multiple virulent factors encoded in babA2, CagA, and vacA genes has been associated with increased risk of GIM, inflammation, and development of gastric cancer.15 The clinical usefulness of genotyping H. pylori strains specifically to survey precancerous gastric lesions remains to be seen because of a lack of sufficient clinical studies. In addition, genotyping H. pylori is not commonly performed as part of clinical practice.
The loss of parietal cells seen in atrophic gastritis due to chronic H. pylori infection has been linked to the development of metaplasia due to possible loss of differentiation-promoting factors. As a result, metaplastic cells emerge that express spasmolytic polypeptide (SP or TFF2); hence, this type of metaplasia is referred to as spasmolytic polypeptide–expressing metaplasia (SPEM). The cellular mechanism that may explain a precursor role of SPEM in the development of GIM remains unknown.14 A second competing theory for the development of GIM is the clonal expansion of stem cells in the gastric isthmus that can lead to dysplasia and cancer development.14
On the basis of histological similarities with small intestinal or colonic epithelium, GIM can be further classified into complete or incomplete intestinal metaplasia.21 Complete intestinal metaplasia most closely resembles small intestinal epithelium with a brush border and goblet cells. Incomplete intestinal metaplasia resembles the colonic epithelium and lacks a brush border. A second classification further classifies GIM into three subtypes: Type I contains nonsecretory absorptive cells and sialomucin secreting goblet cells; type II has few absorptive cells, columnar cells secreting sialomucin, goblet cells secreting mainly sialomucin but some sulphomucin, and presence of Paneth cells; and type III consists of columnar cells secreting predominantly sulphomucin, goblet cells secreting sialomucin or sulphomucin, and absence of Paneth cells.15,22 In this subclassification, type I GIM is known as complete GIM and types II and III as incomplete GIM.23-25
Multiple studies performed outside of the United States have shown a higher progression risk to gastric adenocarcinoma in incomplete intestinal metaplasia, or type III intestinal metaplasia.26-32 Also, the risk of gastric cancer has been demonstrated to be higher among patients with a greater area of metaplasia and extensive intestinal metaplasia, defined as GIM in both the antrum and corpus.33,34 Hence, the extent of the metaplasia determined with mapping biopsies, regardless of the subtype, should also be incorporated into the risk assessment of the patient. Currently, a major limitation in the United States is a standardized method of pathologic reporting including subclassification of incomplete versus complete intestinal metaplasia.
Which patients to screen
Understanding this sequence of carcinogenesis offers a potential window for screening and surveillance. Subsequently, early detection of precancerous mucosal changes would be more amenable for endoscopic submucosal dissection (ESD).35,36 Currently, U.S. society guidelines do not specifically address the management of GIM. The American Society for Gastrointestinal Endoscopy (ASGE) guidelines for management of premalignant and malignant conditions of the stomach recommend surveillance in individuals with a family history of gastric cancer or of high-risk ethnic background but with no specific optimal surveillance interval.37 Also, H. pylori treatment is recommended if identified, but empiric treatment in GIM was felt to be controversial. The AGA recently sought comments on a proposed new guideline for the management of GIM. This guideline should be released after the comment period and help address management of GIM in the United States. In April of 2019, the European Society of Gastrointestinal Endoscopy (ESGE) updated the management of epithelial precancerous conditions and lesions in the stomach (MAPS II) guideline.38 The MAPS II guideline identifies atrophic gastritis and intestinal metaplasia as precancerous lesions. In patients with moderate to marked atrophy or GIM affecting both antral and body mucosa, ESGE recommends endoscopic surveillance with high-definition chromoendoscopy, mapping, and guided biopsies or at least two biopsies taken separately at the lesser and greater curvature of the antrum and body. H. pylori eradication was recommended if the patient tested positive.
Furthermore, MAPS II proposed replacing atrophic gastritis (AG) in the Operative Link on Gastritis Assessment (OLGA) staging by GIM (OLGIM) as it is considered a more reliable predictor of an individual’s gastric neoplasia risk, based on the interobserver agreement kappa value 0.6 for AG versus 0.9 for GIM.39 Five biopsies (two from the antrum, two from the corpus, and one from the incisura angularis) are needed for the OLGA/OLGIM score system to be considered an accurate predictor of this risk.39 This is supported by the early findings of gastric atrophy and GIM in the incisura angularis.23 In addition, for patients with GIM only in either the antrum or the body, a family history of gastric cancer, incomplete GIM, autoimmune gastritis, or persistent H. pylori infection was felt to increase the risk to warrant surveillance every 3 years. In those patients with atrophy or GIM in both the antrum and body with a first-degree relative with gastric cancer, surveillance was recommended every 1-2 years. Patients with any dysplasia and a visible lesion should have staging and resection. With no visible lesion, a follow-up endoscopy should be performed in 6 months with high-grade dysplasia and with low-grade dysplasia a repeat in 12 months. Patients with mild to moderate atrophy in the antrum and no intestinal metaplasia were not felt to warrant any further surveillance. (See Figure 1.)
How to screen
Previous studies have found a poor correlation between the endoscopic determination of gastric atrophy and the histologic diagnosis.42 Several studies also found that gastric cancer was missed on initial endoscopic examinations. Sensitivity of endoscopy to detect gastric cancer has ranged from 77% to 93%.43,44 In the United States, there is a lack of standardized quality indicators for upper endoscopy exams. The ESGE has suggested several performance measures to ensure a quality endoscopy exam, including accurate photo documentation, sufficient procedure time of at least 7 minutes, adherence to biopsy protocols, and low complication rates.45 In Asia, a systematic screening protocol is used for photo documentation, and simple techniques such as adequate air insufflation and irrigation to remove mucus are routinely used to improve the endoscopy exam.46,47 The mean time of an endoscopy exam has also been found to increase the detection of neoplastic lesions, as slow endoscopists – with a mean exam duration of 8.6 ± 4.2 min during upper endoscopy – detected threefold more neoplastic lesions than did fast endoscopists.48
A standardized biopsy approach is also important when screening patients. The updated Sydney protocol has been suggested for mapping the stomach to screen for atrophy and GIM. This protocol recommends two biopsies from the antrum (at the lesser and greater curvature), two from the body (at the lesser and greater curvature), and one from the incisura.23 This biopsy protocol was also suggested in the recent MAPS II update, with the biopsy of the incisura felt to be an additional biopsy left to the discretion of the endoscopist. Notably, abnormal appearing mucosal areas should be biopsied separately from the mapping biopsies.
High-definition endoscopy with virtual chromoendoscopy is felt to be better than white-light endoscopy alone at detecting precancerous gastric lesions.38 (See Figure 2.)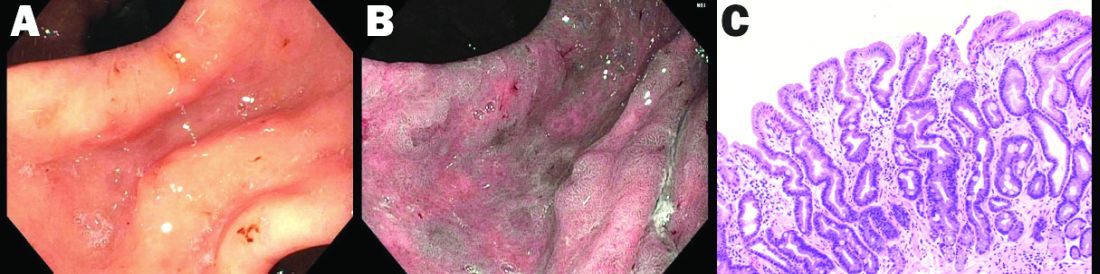
In particular, narrow-band imaging (NBI) has been studied and found to increase the diagnostic yield of GIM and dysplasia compared with white light alone.49 Several studies have shown an increased accuracy for the detection of GIM with magnification NBI.50-52 An unfortunate limitation is the geographic availability of magnification NBI: It is not available in the United States. A multicenter study in Portugal developed a new classification system for the appearance of precancerous lesions with NBI and tested its accuracy in endoscopists with a wide range of NBI experience. An abnormal mucosal pattern that showed light blue crests/regular ridge or a tubulovillous appearance and a regular mucosal pattern was found with GIM. An irregular vascular pattern with a white opaque substance and an absent or irregular mucosal pattern was most often found with dysplasia. Furthermore, the reproducibility of these patterns was high between endoscopists.53 Multiple studies have been performed on additional imaging technologies to enhance the detection of gastric neoplasia; however, these technologies are still investigational and currently not recommended for screening.54-57
Serum pepsinogens have been studied in Europe and Asia as noninvasive indicators of gastric atrophy to determine who should be screened with endoscopy.58 A low serum pepsinogen I level below 70 ng/mL and pepsinogen I/II ratio below 3 has generally been used to detect atrophic gastritis and at-risk populations. However, the studies performed in Europe and Asia used different methods for quantifying pepsinogen levels. Therefore, cutoff values cannot be generalized for all assays and should be validated for the specific tests used.38
Summary
Gastric atrophy and gastric intestinal metaplasia are considered precancerous lesions with an increased risk of development of gastric cancer. H. pylori is a major risk factor for the development of GIM. The extent of GIM as well as the presence of incomplete intestinal metaplasia, or type III intestinal metaplasia has been found to have the highest gastric cancer risk. Currently, in the United States, specific guidelines on endoscopic screening and surveillance for noncardia gastric adenocarcinoma based on histological subtype of GIM, location, and extension are lacking. The ESGE recently updated guidelines that recommend surveillance of patients with extensive atrophy and intestinal metaplasia or with a significant family history. Location and extension of intestinal metaplasia plays a role in increased risk. Screening should include a standardized upper endoscopy approach with high-definition white- light endoscopy and NBI, at least a 7-minute examination, adequate insufflation and cleaning, adequate photo documentation, and a standardized biopsy protocol. Further studies are needed to determine an appropriate surveillance interval and standardized pathology reporting approach as well.
Diana Curras-Martin MD, is an internal medicine resident at Hackensack Meridian Jersey Shore University Medical Center. Susana Gonzalez, MD, is assistant professor of medicine in the division of gastroenterology and hepatology (@WCM_GI), Weill Cornell Medicine, New York Presbyterian Hospital–Cornell.
References
1. Bray F et al. CA Cancer J Clin. 2018;68(6):394-424.
2. Global Burden of Disease Cancer Collaboration et al. JAMA Oncol. 2018;4(11):1553-68.
3. Balakrishnan M et al. Curr Gastroenterol Rep. 2017;19(8):36.
4. Anderson WF et al. J Natl Cancer Inst. 2018;110(6):608-15.
5. Trieu JA et al. Dig Dis Sci. 2019;64(5):1079-88.
6. Lauren P. Acta Pathol Microbiol Scand. 1965;64:31-49.
7. Correa P, Schneider BG. Cancer Epidemiol Biomarkers Prev. 2005;14(8):1865-8.
8. Correa P. Cancer Res. 1992;52(24):6735-40.
9. Correa P, Piazuelo MB. J Dig Dis. 2012;13(1):2-9.
10. Correa P et al. J Natl Cancer Inst. 1970;44(2):297-306.
11. Correa P. Semin Oncol. 1985;12(1):2-10.
12. Rugge M et al. Hum Pathol. 1991;22(10):1002-8.
13. Simko V et al. Bratisl Lek Listy. 2015;116(1):3-8.
14. Giroux V, Rustgi AK. Nat Rev Cancer. 2017;17(10):594-604.
15. Jencks DS et al. Gastroenterol Hepatol (N Y). 2018;14(2):92-101.
16. Amieva M, Peek RM Jr. Gastroenterology. 2016;150(1):64-78.
17. Karimi P et al. Cancer Epidemiol Biomarkers Prev. 2014;23(5):700-13.
18. Hatakeyama M. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93(4):196-219.
19. Tsutsumi R et al. Mol Cell Biol. 2006;26(1):261-76.
20. Kikuchi S et al. Am J Gastroenterol. 1999;94(12):3455-9.
21. Jass JR, Filipe MI. Histopathology. 1980;4(3):271-9.
22. Jass JR, Filipe MI. Histochem J. 1981;13(6):931-9.
23. Dixon MF et al. Am J Surg Pathol. 1996;20(10):1161-81.
24. Kang KP et al. J Gastroenterol Hepatol. 2009;24(1):140-8.
25. Gonzalez CA et al. Int J Cancer. 2010;127(11):2654-60.
26. Filipe MI et al. Gut. 1985;26(12):1319-26.
27. Filipe MI et al. Int J Cancer. 1994;57(3):324-9.
28. Gonzalez CA et al. J Gastroenterol Hepatol. 2016;31(5):953-8.
29. Cassaro M et al. Am J Gastroenterol. 2000;95(6):1431-8.
30. Shao L et al. Int J Cancer. Apr 29. 2018.
31. Stemmermann GN. Cancer. 1994;74(2):556-64.
32. Gonzalez CA et al. Int J Cancer. 2013;133(5):1023-32.
33. Reddy KM et al. Clin Gastroenterol Hepatol. 2016;14(10):1420-5.
34. Tava F et al. Hum Pathol. 2006;37(11):1489-97.
35. Fernandez-Esparrach G et al. Rev Esp Enferm Dig. 2014;106(2):120-32.
36. Ono H et al. Dig Endosc. 2016;28(1):3-15.
37. Evans JA, DeWitt JM. Gastrointest Endosc. 2016;83(1):274.
38. Pimentel-Nunes P et al. Endoscopy. 2019;51(4):365-88.
39. Capelle LG et al. Gastrointest Endosc. 2010;71(7):1150-8.
40. Saumoy M et al. Gastroenterology. 2018;155(3):648-60.
41. Gupta N et al. Gastrointest Endosc. 2011;74(3):610-24 e612.
42. Eshmuratov A et al. Dig Dis Sci. 2010;55(5):1364-75.
43. Nam JH et al. Cancer. 2012;118(20):4953-60.
44. Amin A et al. J R Coll Surg Edinb. 2002;47(5):681-4.
45. Bisschops R et al. United European Gastroenterol J. 2016;4(5):629-56.
46. Uedo N et al. Gastroenterol Clin North Am. 2013;42(2):317-35.
47. Yao K. Ann Gastroenterol. 2013;26(1):11-22.
48. Teh JL et al. Clin Gastroenterol Hepatol. 2015;13(3):480-7 e482.
49. Capelle LG et al. Dig Dis Sci. 2010;55(12):3442-8.
50. Bansal A et al. Gastrointest Endosc. 2008;67(2):210-6.
51. Tahara T et al. Gastrointest Endosc. 2009;70(2):246-53.
52. Uedo N et al. Endoscopy. 2006;38(8):819-24.
53. Pimentel-Nunes P et al. Endoscopy. 2012;44(3):236-46.
54. Kato M et al. Gastrointest Endosc. 2009;70(5):899-906.
55. Nishimura J et al. Gastroenterol Res Pract. 2014;2014:819395.
56. Dohi O et al. Gastrointest Endosc. 2019;89(1):47-57.
57. Osawa H et al. World J Gastrointest Endosc. 2012;4(8):356-61.
58. Pasechnikov V et al. World J Gastroenterol. 2014;20(38):13842-62.
Introduction
Despite a global decline in the incidence of gastric cancer over the past 3 decades, it remains the fifth most commonly diagnosed cancer and the third most common cause of cancer deaths worldwide.1 In the United States it is the fourth most commonly diagnosed GI malignancy, after colorectal, pancreas, and liver cancer. The prevalence remains high in Latin America and Asia, which has implications in the United States because of growing Hispanic and Asian populations.2,3 In recent years, a change in the trend of gastric cancer among non-Hispanic whites has been observed, particularly in women younger than 50 years old.4 5
Etiology
Gastric adenocarcinomas are classified into two subcategories based on location (cardia and noncardia) and histology (intestinal and diffuse types).6,7 Atrophic gastritis and gastric intestinal metaplasia (GIM) are considered precursors of intestinal-type noncardia gastric adenocarcinoma. The Correa cascade is a commonly accepted precancer sequence for noncardia gastric adenocarcinoma that describes mucosal changes from inflammation to atrophy to metaplasia to intraepithelial neoplasia and culminating in carcinoma.8,9 It has been observed that GIM may be the histologic change prior to the development of dysplasia and over 50% of patients with high-grade dysplasia will progress to adenocarcinoma.10-12 In the United States, GIM has the highest prevalence in African Americans, Hispanics, and East Asians, with the overall GIM prevalence regardless of ethnicity reported from 3.05% to 19.2%.5,13
Risk factors and subclassification
Replacement of the foveolar and/or glandular epithelium in the oxyntic and antral mucosa by intestinal epithelium results in GIM. It can be focal when limited to one region of the stomach or extensive when two or more regions are involved.14 The main risk factors for GIM development are Helicobacter pylori infection, tobacco, alcohol consumption, high salt intake, and chronic bile reflux.15,16 Additional risks for developing gastric cancer include older age, certain ethnicities, and male sex.17
CagA strains of H. pylori can promote carcinogenesis by inducing a mitogenic cellular response and downregulating cell adhesion.18,19 Less carcinogenic risk is associated with H. pylori Cag-A negative strains; however, they also have oncogenic potential mediated by expression of babA2 and vacA genes.20 Hence, the combination of multiple virulent factors encoded in babA2, CagA, and vacA genes has been associated with increased risk of GIM, inflammation, and development of gastric cancer.15 The clinical usefulness of genotyping H. pylori strains specifically to survey precancerous gastric lesions remains to be seen because of a lack of sufficient clinical studies. In addition, genotyping H. pylori is not commonly performed as part of clinical practice.
The loss of parietal cells seen in atrophic gastritis due to chronic H. pylori infection has been linked to the development of metaplasia due to possible loss of differentiation-promoting factors. As a result, metaplastic cells emerge that express spasmolytic polypeptide (SP or TFF2); hence, this type of metaplasia is referred to as spasmolytic polypeptide–expressing metaplasia (SPEM). The cellular mechanism that may explain a precursor role of SPEM in the development of GIM remains unknown.14 A second competing theory for the development of GIM is the clonal expansion of stem cells in the gastric isthmus that can lead to dysplasia and cancer development.14
On the basis of histological similarities with small intestinal or colonic epithelium, GIM can be further classified into complete or incomplete intestinal metaplasia.21 Complete intestinal metaplasia most closely resembles small intestinal epithelium with a brush border and goblet cells. Incomplete intestinal metaplasia resembles the colonic epithelium and lacks a brush border. A second classification further classifies GIM into three subtypes: Type I contains nonsecretory absorptive cells and sialomucin secreting goblet cells; type II has few absorptive cells, columnar cells secreting sialomucin, goblet cells secreting mainly sialomucin but some sulphomucin, and presence of Paneth cells; and type III consists of columnar cells secreting predominantly sulphomucin, goblet cells secreting sialomucin or sulphomucin, and absence of Paneth cells.15,22 In this subclassification, type I GIM is known as complete GIM and types II and III as incomplete GIM.23-25
Multiple studies performed outside of the United States have shown a higher progression risk to gastric adenocarcinoma in incomplete intestinal metaplasia, or type III intestinal metaplasia.26-32 Also, the risk of gastric cancer has been demonstrated to be higher among patients with a greater area of metaplasia and extensive intestinal metaplasia, defined as GIM in both the antrum and corpus.33,34 Hence, the extent of the metaplasia determined with mapping biopsies, regardless of the subtype, should also be incorporated into the risk assessment of the patient. Currently, a major limitation in the United States is a standardized method of pathologic reporting including subclassification of incomplete versus complete intestinal metaplasia.
Which patients to screen
Understanding this sequence of carcinogenesis offers a potential window for screening and surveillance. Subsequently, early detection of precancerous mucosal changes would be more amenable for endoscopic submucosal dissection (ESD).35,36 Currently, U.S. society guidelines do not specifically address the management of GIM. The American Society for Gastrointestinal Endoscopy (ASGE) guidelines for management of premalignant and malignant conditions of the stomach recommend surveillance in individuals with a family history of gastric cancer or of high-risk ethnic background but with no specific optimal surveillance interval.37 Also, H. pylori treatment is recommended if identified, but empiric treatment in GIM was felt to be controversial. The AGA recently sought comments on a proposed new guideline for the management of GIM. This guideline should be released after the comment period and help address management of GIM in the United States. In April of 2019, the European Society of Gastrointestinal Endoscopy (ESGE) updated the management of epithelial precancerous conditions and lesions in the stomach (MAPS II) guideline.38 The MAPS II guideline identifies atrophic gastritis and intestinal metaplasia as precancerous lesions. In patients with moderate to marked atrophy or GIM affecting both antral and body mucosa, ESGE recommends endoscopic surveillance with high-definition chromoendoscopy, mapping, and guided biopsies or at least two biopsies taken separately at the lesser and greater curvature of the antrum and body. H. pylori eradication was recommended if the patient tested positive.
Furthermore, MAPS II proposed replacing atrophic gastritis (AG) in the Operative Link on Gastritis Assessment (OLGA) staging by GIM (OLGIM) as it is considered a more reliable predictor of an individual’s gastric neoplasia risk, based on the interobserver agreement kappa value 0.6 for AG versus 0.9 for GIM.39 Five biopsies (two from the antrum, two from the corpus, and one from the incisura angularis) are needed for the OLGA/OLGIM score system to be considered an accurate predictor of this risk.39 This is supported by the early findings of gastric atrophy and GIM in the incisura angularis.23 In addition, for patients with GIM only in either the antrum or the body, a family history of gastric cancer, incomplete GIM, autoimmune gastritis, or persistent H. pylori infection was felt to increase the risk to warrant surveillance every 3 years. In those patients with atrophy or GIM in both the antrum and body with a first-degree relative with gastric cancer, surveillance was recommended every 1-2 years. Patients with any dysplasia and a visible lesion should have staging and resection. With no visible lesion, a follow-up endoscopy should be performed in 6 months with high-grade dysplasia and with low-grade dysplasia a repeat in 12 months. Patients with mild to moderate atrophy in the antrum and no intestinal metaplasia were not felt to warrant any further surveillance. (See Figure 1.)
How to screen
Previous studies have found a poor correlation between the endoscopic determination of gastric atrophy and the histologic diagnosis.42 Several studies also found that gastric cancer was missed on initial endoscopic examinations. Sensitivity of endoscopy to detect gastric cancer has ranged from 77% to 93%.43,44 In the United States, there is a lack of standardized quality indicators for upper endoscopy exams. The ESGE has suggested several performance measures to ensure a quality endoscopy exam, including accurate photo documentation, sufficient procedure time of at least 7 minutes, adherence to biopsy protocols, and low complication rates.45 In Asia, a systematic screening protocol is used for photo documentation, and simple techniques such as adequate air insufflation and irrigation to remove mucus are routinely used to improve the endoscopy exam.46,47 The mean time of an endoscopy exam has also been found to increase the detection of neoplastic lesions, as slow endoscopists – with a mean exam duration of 8.6 ± 4.2 min during upper endoscopy – detected threefold more neoplastic lesions than did fast endoscopists.48
A standardized biopsy approach is also important when screening patients. The updated Sydney protocol has been suggested for mapping the stomach to screen for atrophy and GIM. This protocol recommends two biopsies from the antrum (at the lesser and greater curvature), two from the body (at the lesser and greater curvature), and one from the incisura.23 This biopsy protocol was also suggested in the recent MAPS II update, with the biopsy of the incisura felt to be an additional biopsy left to the discretion of the endoscopist. Notably, abnormal appearing mucosal areas should be biopsied separately from the mapping biopsies.
High-definition endoscopy with virtual chromoendoscopy is felt to be better than white-light endoscopy alone at detecting precancerous gastric lesions.38 (See Figure 2.)
In particular, narrow-band imaging (NBI) has been studied and found to increase the diagnostic yield of GIM and dysplasia compared with white light alone.49 Several studies have shown an increased accuracy for the detection of GIM with magnification NBI.50-52 An unfortunate limitation is the geographic availability of magnification NBI: It is not available in the United States. A multicenter study in Portugal developed a new classification system for the appearance of precancerous lesions with NBI and tested its accuracy in endoscopists with a wide range of NBI experience. An abnormal mucosal pattern that showed light blue crests/regular ridge or a tubulovillous appearance and a regular mucosal pattern was found with GIM. An irregular vascular pattern with a white opaque substance and an absent or irregular mucosal pattern was most often found with dysplasia. Furthermore, the reproducibility of these patterns was high between endoscopists.53 Multiple studies have been performed on additional imaging technologies to enhance the detection of gastric neoplasia; however, these technologies are still investigational and currently not recommended for screening.54-57
Serum pepsinogens have been studied in Europe and Asia as noninvasive indicators of gastric atrophy to determine who should be screened with endoscopy.58 A low serum pepsinogen I level below 70 ng/mL and pepsinogen I/II ratio below 3 has generally been used to detect atrophic gastritis and at-risk populations. However, the studies performed in Europe and Asia used different methods for quantifying pepsinogen levels. Therefore, cutoff values cannot be generalized for all assays and should be validated for the specific tests used.38
Summary
Gastric atrophy and gastric intestinal metaplasia are considered precancerous lesions with an increased risk of development of gastric cancer. H. pylori is a major risk factor for the development of GIM. The extent of GIM as well as the presence of incomplete intestinal metaplasia, or type III intestinal metaplasia has been found to have the highest gastric cancer risk. Currently, in the United States, specific guidelines on endoscopic screening and surveillance for noncardia gastric adenocarcinoma based on histological subtype of GIM, location, and extension are lacking. The ESGE recently updated guidelines that recommend surveillance of patients with extensive atrophy and intestinal metaplasia or with a significant family history. Location and extension of intestinal metaplasia plays a role in increased risk. Screening should include a standardized upper endoscopy approach with high-definition white- light endoscopy and NBI, at least a 7-minute examination, adequate insufflation and cleaning, adequate photo documentation, and a standardized biopsy protocol. Further studies are needed to determine an appropriate surveillance interval and standardized pathology reporting approach as well.
Diana Curras-Martin MD, is an internal medicine resident at Hackensack Meridian Jersey Shore University Medical Center. Susana Gonzalez, MD, is assistant professor of medicine in the division of gastroenterology and hepatology (@WCM_GI), Weill Cornell Medicine, New York Presbyterian Hospital–Cornell.
References
1. Bray F et al. CA Cancer J Clin. 2018;68(6):394-424.
2. Global Burden of Disease Cancer Collaboration et al. JAMA Oncol. 2018;4(11):1553-68.
3. Balakrishnan M et al. Curr Gastroenterol Rep. 2017;19(8):36.
4. Anderson WF et al. J Natl Cancer Inst. 2018;110(6):608-15.
5. Trieu JA et al. Dig Dis Sci. 2019;64(5):1079-88.
6. Lauren P. Acta Pathol Microbiol Scand. 1965;64:31-49.
7. Correa P, Schneider BG. Cancer Epidemiol Biomarkers Prev. 2005;14(8):1865-8.
8. Correa P. Cancer Res. 1992;52(24):6735-40.
9. Correa P, Piazuelo MB. J Dig Dis. 2012;13(1):2-9.
10. Correa P et al. J Natl Cancer Inst. 1970;44(2):297-306.
11. Correa P. Semin Oncol. 1985;12(1):2-10.
12. Rugge M et al. Hum Pathol. 1991;22(10):1002-8.
13. Simko V et al. Bratisl Lek Listy. 2015;116(1):3-8.
14. Giroux V, Rustgi AK. Nat Rev Cancer. 2017;17(10):594-604.
15. Jencks DS et al. Gastroenterol Hepatol (N Y). 2018;14(2):92-101.
16. Amieva M, Peek RM Jr. Gastroenterology. 2016;150(1):64-78.
17. Karimi P et al. Cancer Epidemiol Biomarkers Prev. 2014;23(5):700-13.
18. Hatakeyama M. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93(4):196-219.
19. Tsutsumi R et al. Mol Cell Biol. 2006;26(1):261-76.
20. Kikuchi S et al. Am J Gastroenterol. 1999;94(12):3455-9.
21. Jass JR, Filipe MI. Histopathology. 1980;4(3):271-9.
22. Jass JR, Filipe MI. Histochem J. 1981;13(6):931-9.
23. Dixon MF et al. Am J Surg Pathol. 1996;20(10):1161-81.
24. Kang KP et al. J Gastroenterol Hepatol. 2009;24(1):140-8.
25. Gonzalez CA et al. Int J Cancer. 2010;127(11):2654-60.
26. Filipe MI et al. Gut. 1985;26(12):1319-26.
27. Filipe MI et al. Int J Cancer. 1994;57(3):324-9.
28. Gonzalez CA et al. J Gastroenterol Hepatol. 2016;31(5):953-8.
29. Cassaro M et al. Am J Gastroenterol. 2000;95(6):1431-8.
30. Shao L et al. Int J Cancer. Apr 29. 2018.
31. Stemmermann GN. Cancer. 1994;74(2):556-64.
32. Gonzalez CA et al. Int J Cancer. 2013;133(5):1023-32.
33. Reddy KM et al. Clin Gastroenterol Hepatol. 2016;14(10):1420-5.
34. Tava F et al. Hum Pathol. 2006;37(11):1489-97.
35. Fernandez-Esparrach G et al. Rev Esp Enferm Dig. 2014;106(2):120-32.
36. Ono H et al. Dig Endosc. 2016;28(1):3-15.
37. Evans JA, DeWitt JM. Gastrointest Endosc. 2016;83(1):274.
38. Pimentel-Nunes P et al. Endoscopy. 2019;51(4):365-88.
39. Capelle LG et al. Gastrointest Endosc. 2010;71(7):1150-8.
40. Saumoy M et al. Gastroenterology. 2018;155(3):648-60.
41. Gupta N et al. Gastrointest Endosc. 2011;74(3):610-24 e612.
42. Eshmuratov A et al. Dig Dis Sci. 2010;55(5):1364-75.
43. Nam JH et al. Cancer. 2012;118(20):4953-60.
44. Amin A et al. J R Coll Surg Edinb. 2002;47(5):681-4.
45. Bisschops R et al. United European Gastroenterol J. 2016;4(5):629-56.
46. Uedo N et al. Gastroenterol Clin North Am. 2013;42(2):317-35.
47. Yao K. Ann Gastroenterol. 2013;26(1):11-22.
48. Teh JL et al. Clin Gastroenterol Hepatol. 2015;13(3):480-7 e482.
49. Capelle LG et al. Dig Dis Sci. 2010;55(12):3442-8.
50. Bansal A et al. Gastrointest Endosc. 2008;67(2):210-6.
51. Tahara T et al. Gastrointest Endosc. 2009;70(2):246-53.
52. Uedo N et al. Endoscopy. 2006;38(8):819-24.
53. Pimentel-Nunes P et al. Endoscopy. 2012;44(3):236-46.
54. Kato M et al. Gastrointest Endosc. 2009;70(5):899-906.
55. Nishimura J et al. Gastroenterol Res Pract. 2014;2014:819395.
56. Dohi O et al. Gastrointest Endosc. 2019;89(1):47-57.
57. Osawa H et al. World J Gastrointest Endosc. 2012;4(8):356-61.
58. Pasechnikov V et al. World J Gastroenterol. 2014;20(38):13842-62.
Introduction
Despite a global decline in the incidence of gastric cancer over the past 3 decades, it remains the fifth most commonly diagnosed cancer and the third most common cause of cancer deaths worldwide.1 In the United States it is the fourth most commonly diagnosed GI malignancy, after colorectal, pancreas, and liver cancer. The prevalence remains high in Latin America and Asia, which has implications in the United States because of growing Hispanic and Asian populations.2,3 In recent years, a change in the trend of gastric cancer among non-Hispanic whites has been observed, particularly in women younger than 50 years old.4 5
Etiology
Gastric adenocarcinomas are classified into two subcategories based on location (cardia and noncardia) and histology (intestinal and diffuse types).6,7 Atrophic gastritis and gastric intestinal metaplasia (GIM) are considered precursors of intestinal-type noncardia gastric adenocarcinoma. The Correa cascade is a commonly accepted precancer sequence for noncardia gastric adenocarcinoma that describes mucosal changes from inflammation to atrophy to metaplasia to intraepithelial neoplasia and culminating in carcinoma.8,9 It has been observed that GIM may be the histologic change prior to the development of dysplasia and over 50% of patients with high-grade dysplasia will progress to adenocarcinoma.10-12 In the United States, GIM has the highest prevalence in African Americans, Hispanics, and East Asians, with the overall GIM prevalence regardless of ethnicity reported from 3.05% to 19.2%.5,13
Risk factors and subclassification
Replacement of the foveolar and/or glandular epithelium in the oxyntic and antral mucosa by intestinal epithelium results in GIM. It can be focal when limited to one region of the stomach or extensive when two or more regions are involved.14 The main risk factors for GIM development are Helicobacter pylori infection, tobacco, alcohol consumption, high salt intake, and chronic bile reflux.15,16 Additional risks for developing gastric cancer include older age, certain ethnicities, and male sex.17
CagA strains of H. pylori can promote carcinogenesis by inducing a mitogenic cellular response and downregulating cell adhesion.18,19 Less carcinogenic risk is associated with H. pylori Cag-A negative strains; however, they also have oncogenic potential mediated by expression of babA2 and vacA genes.20 Hence, the combination of multiple virulent factors encoded in babA2, CagA, and vacA genes has been associated with increased risk of GIM, inflammation, and development of gastric cancer.15 The clinical usefulness of genotyping H. pylori strains specifically to survey precancerous gastric lesions remains to be seen because of a lack of sufficient clinical studies. In addition, genotyping H. pylori is not commonly performed as part of clinical practice.
The loss of parietal cells seen in atrophic gastritis due to chronic H. pylori infection has been linked to the development of metaplasia due to possible loss of differentiation-promoting factors. As a result, metaplastic cells emerge that express spasmolytic polypeptide (SP or TFF2); hence, this type of metaplasia is referred to as spasmolytic polypeptide–expressing metaplasia (SPEM). The cellular mechanism that may explain a precursor role of SPEM in the development of GIM remains unknown.14 A second competing theory for the development of GIM is the clonal expansion of stem cells in the gastric isthmus that can lead to dysplasia and cancer development.14
On the basis of histological similarities with small intestinal or colonic epithelium, GIM can be further classified into complete or incomplete intestinal metaplasia.21 Complete intestinal metaplasia most closely resembles small intestinal epithelium with a brush border and goblet cells. Incomplete intestinal metaplasia resembles the colonic epithelium and lacks a brush border. A second classification further classifies GIM into three subtypes: Type I contains nonsecretory absorptive cells and sialomucin secreting goblet cells; type II has few absorptive cells, columnar cells secreting sialomucin, goblet cells secreting mainly sialomucin but some sulphomucin, and presence of Paneth cells; and type III consists of columnar cells secreting predominantly sulphomucin, goblet cells secreting sialomucin or sulphomucin, and absence of Paneth cells.15,22 In this subclassification, type I GIM is known as complete GIM and types II and III as incomplete GIM.23-25
Multiple studies performed outside of the United States have shown a higher progression risk to gastric adenocarcinoma in incomplete intestinal metaplasia, or type III intestinal metaplasia.26-32 Also, the risk of gastric cancer has been demonstrated to be higher among patients with a greater area of metaplasia and extensive intestinal metaplasia, defined as GIM in both the antrum and corpus.33,34 Hence, the extent of the metaplasia determined with mapping biopsies, regardless of the subtype, should also be incorporated into the risk assessment of the patient. Currently, a major limitation in the United States is a standardized method of pathologic reporting including subclassification of incomplete versus complete intestinal metaplasia.
Which patients to screen
Understanding this sequence of carcinogenesis offers a potential window for screening and surveillance. Subsequently, early detection of precancerous mucosal changes would be more amenable for endoscopic submucosal dissection (ESD).35,36 Currently, U.S. society guidelines do not specifically address the management of GIM. The American Society for Gastrointestinal Endoscopy (ASGE) guidelines for management of premalignant and malignant conditions of the stomach recommend surveillance in individuals with a family history of gastric cancer or of high-risk ethnic background but with no specific optimal surveillance interval.37 Also, H. pylori treatment is recommended if identified, but empiric treatment in GIM was felt to be controversial. The AGA recently sought comments on a proposed new guideline for the management of GIM. This guideline should be released after the comment period and help address management of GIM in the United States. In April of 2019, the European Society of Gastrointestinal Endoscopy (ESGE) updated the management of epithelial precancerous conditions and lesions in the stomach (MAPS II) guideline.38 The MAPS II guideline identifies atrophic gastritis and intestinal metaplasia as precancerous lesions. In patients with moderate to marked atrophy or GIM affecting both antral and body mucosa, ESGE recommends endoscopic surveillance with high-definition chromoendoscopy, mapping, and guided biopsies or at least two biopsies taken separately at the lesser and greater curvature of the antrum and body. H. pylori eradication was recommended if the patient tested positive.
Furthermore, MAPS II proposed replacing atrophic gastritis (AG) in the Operative Link on Gastritis Assessment (OLGA) staging by GIM (OLGIM) as it is considered a more reliable predictor of an individual’s gastric neoplasia risk, based on the interobserver agreement kappa value 0.6 for AG versus 0.9 for GIM.39 Five biopsies (two from the antrum, two from the corpus, and one from the incisura angularis) are needed for the OLGA/OLGIM score system to be considered an accurate predictor of this risk.39 This is supported by the early findings of gastric atrophy and GIM in the incisura angularis.23 In addition, for patients with GIM only in either the antrum or the body, a family history of gastric cancer, incomplete GIM, autoimmune gastritis, or persistent H. pylori infection was felt to increase the risk to warrant surveillance every 3 years. In those patients with atrophy or GIM in both the antrum and body with a first-degree relative with gastric cancer, surveillance was recommended every 1-2 years. Patients with any dysplasia and a visible lesion should have staging and resection. With no visible lesion, a follow-up endoscopy should be performed in 6 months with high-grade dysplasia and with low-grade dysplasia a repeat in 12 months. Patients with mild to moderate atrophy in the antrum and no intestinal metaplasia were not felt to warrant any further surveillance. (See Figure 1.)
How to screen
Previous studies have found a poor correlation between the endoscopic determination of gastric atrophy and the histologic diagnosis.42 Several studies also found that gastric cancer was missed on initial endoscopic examinations. Sensitivity of endoscopy to detect gastric cancer has ranged from 77% to 93%.43,44 In the United States, there is a lack of standardized quality indicators for upper endoscopy exams. The ESGE has suggested several performance measures to ensure a quality endoscopy exam, including accurate photo documentation, sufficient procedure time of at least 7 minutes, adherence to biopsy protocols, and low complication rates.45 In Asia, a systematic screening protocol is used for photo documentation, and simple techniques such as adequate air insufflation and irrigation to remove mucus are routinely used to improve the endoscopy exam.46,47 The mean time of an endoscopy exam has also been found to increase the detection of neoplastic lesions, as slow endoscopists – with a mean exam duration of 8.6 ± 4.2 min during upper endoscopy – detected threefold more neoplastic lesions than did fast endoscopists.48
A standardized biopsy approach is also important when screening patients. The updated Sydney protocol has been suggested for mapping the stomach to screen for atrophy and GIM. This protocol recommends two biopsies from the antrum (at the lesser and greater curvature), two from the body (at the lesser and greater curvature), and one from the incisura.23 This biopsy protocol was also suggested in the recent MAPS II update, with the biopsy of the incisura felt to be an additional biopsy left to the discretion of the endoscopist. Notably, abnormal appearing mucosal areas should be biopsied separately from the mapping biopsies.
High-definition endoscopy with virtual chromoendoscopy is felt to be better than white-light endoscopy alone at detecting precancerous gastric lesions.38 (See Figure 2.)
In particular, narrow-band imaging (NBI) has been studied and found to increase the diagnostic yield of GIM and dysplasia compared with white light alone.49 Several studies have shown an increased accuracy for the detection of GIM with magnification NBI.50-52 An unfortunate limitation is the geographic availability of magnification NBI: It is not available in the United States. A multicenter study in Portugal developed a new classification system for the appearance of precancerous lesions with NBI and tested its accuracy in endoscopists with a wide range of NBI experience. An abnormal mucosal pattern that showed light blue crests/regular ridge or a tubulovillous appearance and a regular mucosal pattern was found with GIM. An irregular vascular pattern with a white opaque substance and an absent or irregular mucosal pattern was most often found with dysplasia. Furthermore, the reproducibility of these patterns was high between endoscopists.53 Multiple studies have been performed on additional imaging technologies to enhance the detection of gastric neoplasia; however, these technologies are still investigational and currently not recommended for screening.54-57
Serum pepsinogens have been studied in Europe and Asia as noninvasive indicators of gastric atrophy to determine who should be screened with endoscopy.58 A low serum pepsinogen I level below 70 ng/mL and pepsinogen I/II ratio below 3 has generally been used to detect atrophic gastritis and at-risk populations. However, the studies performed in Europe and Asia used different methods for quantifying pepsinogen levels. Therefore, cutoff values cannot be generalized for all assays and should be validated for the specific tests used.38
Summary
Gastric atrophy and gastric intestinal metaplasia are considered precancerous lesions with an increased risk of development of gastric cancer. H. pylori is a major risk factor for the development of GIM. The extent of GIM as well as the presence of incomplete intestinal metaplasia, or type III intestinal metaplasia has been found to have the highest gastric cancer risk. Currently, in the United States, specific guidelines on endoscopic screening and surveillance for noncardia gastric adenocarcinoma based on histological subtype of GIM, location, and extension are lacking. The ESGE recently updated guidelines that recommend surveillance of patients with extensive atrophy and intestinal metaplasia or with a significant family history. Location and extension of intestinal metaplasia plays a role in increased risk. Screening should include a standardized upper endoscopy approach with high-definition white- light endoscopy and NBI, at least a 7-minute examination, adequate insufflation and cleaning, adequate photo documentation, and a standardized biopsy protocol. Further studies are needed to determine an appropriate surveillance interval and standardized pathology reporting approach as well.
Diana Curras-Martin MD, is an internal medicine resident at Hackensack Meridian Jersey Shore University Medical Center. Susana Gonzalez, MD, is assistant professor of medicine in the division of gastroenterology and hepatology (@WCM_GI), Weill Cornell Medicine, New York Presbyterian Hospital–Cornell.
References
1. Bray F et al. CA Cancer J Clin. 2018;68(6):394-424.
2. Global Burden of Disease Cancer Collaboration et al. JAMA Oncol. 2018;4(11):1553-68.
3. Balakrishnan M et al. Curr Gastroenterol Rep. 2017;19(8):36.
4. Anderson WF et al. J Natl Cancer Inst. 2018;110(6):608-15.
5. Trieu JA et al. Dig Dis Sci. 2019;64(5):1079-88.
6. Lauren P. Acta Pathol Microbiol Scand. 1965;64:31-49.
7. Correa P, Schneider BG. Cancer Epidemiol Biomarkers Prev. 2005;14(8):1865-8.
8. Correa P. Cancer Res. 1992;52(24):6735-40.
9. Correa P, Piazuelo MB. J Dig Dis. 2012;13(1):2-9.
10. Correa P et al. J Natl Cancer Inst. 1970;44(2):297-306.
11. Correa P. Semin Oncol. 1985;12(1):2-10.
12. Rugge M et al. Hum Pathol. 1991;22(10):1002-8.
13. Simko V et al. Bratisl Lek Listy. 2015;116(1):3-8.
14. Giroux V, Rustgi AK. Nat Rev Cancer. 2017;17(10):594-604.
15. Jencks DS et al. Gastroenterol Hepatol (N Y). 2018;14(2):92-101.
16. Amieva M, Peek RM Jr. Gastroenterology. 2016;150(1):64-78.
17. Karimi P et al. Cancer Epidemiol Biomarkers Prev. 2014;23(5):700-13.
18. Hatakeyama M. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93(4):196-219.
19. Tsutsumi R et al. Mol Cell Biol. 2006;26(1):261-76.
20. Kikuchi S et al. Am J Gastroenterol. 1999;94(12):3455-9.
21. Jass JR, Filipe MI. Histopathology. 1980;4(3):271-9.
22. Jass JR, Filipe MI. Histochem J. 1981;13(6):931-9.
23. Dixon MF et al. Am J Surg Pathol. 1996;20(10):1161-81.
24. Kang KP et al. J Gastroenterol Hepatol. 2009;24(1):140-8.
25. Gonzalez CA et al. Int J Cancer. 2010;127(11):2654-60.
26. Filipe MI et al. Gut. 1985;26(12):1319-26.
27. Filipe MI et al. Int J Cancer. 1994;57(3):324-9.
28. Gonzalez CA et al. J Gastroenterol Hepatol. 2016;31(5):953-8.
29. Cassaro M et al. Am J Gastroenterol. 2000;95(6):1431-8.
30. Shao L et al. Int J Cancer. Apr 29. 2018.
31. Stemmermann GN. Cancer. 1994;74(2):556-64.
32. Gonzalez CA et al. Int J Cancer. 2013;133(5):1023-32.
33. Reddy KM et al. Clin Gastroenterol Hepatol. 2016;14(10):1420-5.
34. Tava F et al. Hum Pathol. 2006;37(11):1489-97.
35. Fernandez-Esparrach G et al. Rev Esp Enferm Dig. 2014;106(2):120-32.
36. Ono H et al. Dig Endosc. 2016;28(1):3-15.
37. Evans JA, DeWitt JM. Gastrointest Endosc. 2016;83(1):274.
38. Pimentel-Nunes P et al. Endoscopy. 2019;51(4):365-88.
39. Capelle LG et al. Gastrointest Endosc. 2010;71(7):1150-8.
40. Saumoy M et al. Gastroenterology. 2018;155(3):648-60.
41. Gupta N et al. Gastrointest Endosc. 2011;74(3):610-24 e612.
42. Eshmuratov A et al. Dig Dis Sci. 2010;55(5):1364-75.
43. Nam JH et al. Cancer. 2012;118(20):4953-60.
44. Amin A et al. J R Coll Surg Edinb. 2002;47(5):681-4.
45. Bisschops R et al. United European Gastroenterol J. 2016;4(5):629-56.
46. Uedo N et al. Gastroenterol Clin North Am. 2013;42(2):317-35.
47. Yao K. Ann Gastroenterol. 2013;26(1):11-22.
48. Teh JL et al. Clin Gastroenterol Hepatol. 2015;13(3):480-7 e482.
49. Capelle LG et al. Dig Dis Sci. 2010;55(12):3442-8.
50. Bansal A et al. Gastrointest Endosc. 2008;67(2):210-6.
51. Tahara T et al. Gastrointest Endosc. 2009;70(2):246-53.
52. Uedo N et al. Endoscopy. 2006;38(8):819-24.
53. Pimentel-Nunes P et al. Endoscopy. 2012;44(3):236-46.
54. Kato M et al. Gastrointest Endosc. 2009;70(5):899-906.
55. Nishimura J et al. Gastroenterol Res Pract. 2014;2014:819395.
56. Dohi O et al. Gastrointest Endosc. 2019;89(1):47-57.
57. Osawa H et al. World J Gastrointest Endosc. 2012;4(8):356-61.
58. Pasechnikov V et al. World J Gastroenterol. 2014;20(38):13842-62.