User login
The Third Time's the Charm
A 58‐year old woman was brought to the emergency department with confusion. Her husband stated that for several hours she had been drifting in and out at home, and that he had to shout to get her attention. He described no seizure activity, weakness, incontinence, or difficulty speaking, and had noted no complaints of headache, fevers, chest pain, shortness of breath, or gastrointestinal complaints.
Altered mental status in a middle‐aged woman can result from a diverse set of etiologies. A key distinction in the neurological examination will be to assure that the complaint of confusion is accurate as opposed to aphasia; the former is usually indicative of diffuse cerebral dysfunction while the latter suggests a focal lesion in the dominant hemisphere.
The acuity of the change in mental status is important, as are the fluctuations described by the husband. Unwitnessed or nonconvulsive seizure activity can present this way. Toxic/metabolic etiologies, infectious and inflammatory disorders of the central nervous system (CNS), and vascular diseases are also important considerations. Although stroke does not typically present with global encephalopathy, intermittent large vessel occlusion, especially in the posterior circulation, can disrupt cognition in this manner. Following a physical examination, initial workup should focus on toxic/metabolic etiologies, followed rapidly by head imaging if no cause is identified.
Her past medical history was notable for type 2 diabetes mellitus, coronary artery disease, hyperlipidemia, and an unspecified seizure disorder, which according to her husband was diagnosed during a recent hospitalization for a similar presentation. She also had a remote history of venous thromboembolism and antithrombin‐III deficiency. She was unemployed, lived with her husband, and spent most of her time at home. She never smoked, and rarely drank alcohol. Her family history was unobtainable, and her husband denied that she used any illicit drugs. Her medications included pioglitazone, aspirin, simvastatin, pregabalin, ferrous sulfate, levetiracetam, warfarin, and magnesium oxide, and she was allergic to sulfa.
While the differential diagnosis remains broad, 3 elements of the history are potentially relevant. The history of epilepsy based on a similar prior presentation increases the likelihood that the current spell is ictal in nature; examination of previous records would be important in order to document whether these spells have indeed been proven to be epileptic, as many conditions can mimic seizures. Given the history of venous thromboembolism and hypercoagulability, one must consider cerebral venous sinus thrombosis, which can present with global neurologic dysfunction and seizures. Prompt identification, usually via computed tomography (CT) or magnetic resonance angiography, is vital, because anticoagulation can mitigate this potentially life‐threatening illness. Finally, although many medications can cause encephalopathy in overdose, levetiracetam has well‐described cognitive side effects even at usual doses, including encephalopathy, irritability, and depression.
The records from that recent hospitalization remarked that she had presented confused and stuporous. Her potassium had been 2.7 mmol/L, international normalized ration (INR) 3.4, and hemoglobin 8 g/dL; other routine laboratory studies were normal. CT and magnetic resonance imaging (MRI) of the brain had been negative, and electroencephalogram (EEG) reportedly was performed but specific results were unknown. She was discharged alert and oriented 1 week prior to the current presentation on the above medications, including levetiracetam for this newly‐diagnosed seizure disorder.
Previous records confirm that the current presentation is that of a relapsing acute alteration in mental status. Regardless of the EEG findings or response to antiepileptic medications, a seizure disorder should remain a primary consideration, although relapsing inflammatory, toxic/metabolic conditions, and, rarely, vascular disorders can also present in this manner.
The neurologic manifestations of hypokalemia are usually peripheral in nature, including periodic paralysis; confusion accompanying hypokalemia is usually not a result of the low potassium itself but rather due to an underlying toxic or endocrinologic cause. Various causes of anemia can lead to mental status changes; the mean corpuscular volume (MCV) will be particularly helpful given known associations between megaloblastic anemia and confusional states.
On examination, she appeared to be in good health and in no distress. She was afebrile. Her blood pressure was 93/57, pulse 90 beats per minute, respiratory rate 16 per minute, and room air oxygen saturation 100%. She was oriented to her surroundings, but slow in her responses to questioning. There were no cranial nerve, motor, or sensory deficits, or abnormal reflexes or movements. Examination of the head, skin, chest, cardiovascular system, abdomen, and extremities was normal. Serum sodium was 136 mmol/L, creatinine 1.2 mg/dL, calcium 9.3 mg/dL, and glucose 81 mg/dL; other routine blood chemistries were normal. Her white blood cell (WBC) count was 7100/L, hemoglobin 9.2 g/dL with normal MCV, and platelet count 275,000/L. INR was 3.4, and liver function tests were normal. CT of the brain demonstrated no evidence of acute pathology.
Given that her laboratory results (aside from the hemoglobin) and CT were essentially normal, the most common etiology of a recurrent encephalopathy would be a toxic exposure including drugs, alcohol, and environmental toxins or poisons. A comprehensive serum drug screen, including heavy metals, could follow a basic urinary screen for drugs of abuse; specific etiologies may be suggested by patterns of injury seen on MRI such as those seen with carbon monoxide or methanol exposure. Other recurrent metabolic processes include the porphyrias and relapsing inflammatory disorders, which could be entertained if further diagnostics are unrevealing.
An EEG is warranted at this point and is a test that is underutilized in the workup of altered mental status. Patients who have a spell and do not quickly awaken should be considered to be in nonconvulsive status epilepticus until proven otherwise. This can be easily identified on the EEG and is an important entity to recognize quickly. Additional findings on EEG may suggest focal cerebral dysfunction (such as that following a seizure or acute unilateral injury), diffuse encephalopathy (eg, triphasic waves), or fairly specific diagnoses (eg, periodic lateralized epileptiform discharges from the temporal lobes in suspected herpes simplex meningoencephalitis). While the CT of the brain is a reasonable initial screen, MRI is more sensitive for structural disease and should be obtained if no etiology is rapidly identified.
Finally, acute infectious etiologies such as abscess, encephalitis, or meningoencephalitis need to be excluded via lumbar puncture. Spinal fluid examination also can be helpful in the consideration of inflammatory and autoimmune disorders.
Over the next several hours, while still in the emergency department, she became increasingly obtunded, to the point that she was unresponsive to all stimuli. No seizure activity was witnessed, her vital signs were unchanged, and no medications had been administered. She was urgently transferred to a tertiary care center, where, at the time of arrival, she was obtunded and nonverbal, and opened her eyes only to noxious stimuli. She would withdraw all 4 extremities in response to pain. Pupils were 2 mm and symmetrically reactive. Corneal reflexes were normal, and her gag reflex was diminished. Motor tone was decreased in all 4 extremities. No fasciculations were noted. Deep tendon reflexes were present but symmetrically diminished throughout, and Babinski testing demonstrated a withdrawal response bilaterally.
Coma is a state of profound unconsciousness where the patient is unarousable and unaware of her surroundings. Coma can result either from bihemispheric dysfunction or diffuse injury to the reticular activating system in the brainstem, and the physical examination should focus on distinguishing between these 2 sites. Because the nuclei of cranial nerves III through XII (excepting XI) reside in the brainstem, the coma examination emphasizes testing the cranial nerves; although all cranial nerves are not tested in this patient, the ones that are appear to be normal, making bihemispheric dysfunction most likely. Bihemispheric coma most commonly results from diffuse toxic or metabolic etiologies such as intoxication or hepatic encephalopathy, but it can also be caused by bilateral structural lesions (including the bilateral thalami) or ongoing seizure activity.
Although an EEG remains the key test in this patient given her past history and an MRI would prove extremely useful, her deterioration warrants a workup for CNS infection. Since the head CT was negative, it would be prudent to proceed with urgent lumbar puncture (although it should never be performed in a patient with significant coagulopathy due to risks of hemorrhage leading to spinal cord injury). She should be covered empirically with broad spectrum meningeal‐dose antibiotics, including acyclovir, until the results of the spinal fluid examination are known, given that bacterial meningitis and herpes meningoencephalitis carry a high morbidity and mortality if not treated promptly.
Routine blood tests were similar to her labs at the referring emergency room. Ammonia level was 10 mol/L. Urine toxicology screen was negative, and blood tests for ethanol, salicylates, lithium, and acetaminophen were negative. Chest X‐ray and urinalysis were normal, and electrocardiogram was notable only for a sinus tachycardia. Cultures of the blood were obtained and the patient was admitted to the intensive care unit.
Levetiracetam, vancomycin, piperacillin‐tazobactam, and acyclovir were initiated. A lumbar puncture was performed without reversing the anticoagulation, and the procedure was traumatic. The cerebrospinal fluid was bloody, with a clear supernatant. Cell count demonstrated a red blood cell (RBC) count of 1250/L and a WBC count of 9/L, with a WBC differential of 42% neutrophils, 48% lymphocytes, and 8% monocytes. The cerebrospinal fluid (CSF) glucose was 62 mg/dL (with a serum glucose of 74 mg/dL) and protein 41 mg/dL. The CSF Gram stain demonstrated no organisms, and fluid was sent for routine culture and polymerase chain reaction (PCR) to detect herpes simplex virus (HSV). A neurology consultation was urgently requested.
As mentioned, it would have been more appropriate to reverse the patient's anticoagulation prior to lumbar puncture. The absence of xanthochromia suggests that the RBCs seen in the sample were introduced at the time of the lumbar puncture, arguing against a hemorrhagic disorder of the CNS (occasionally seen with herpes simplex encephalitis) or spinal fluid (eg, subarachnoid hemorrhage).
A reasonable rule of thumb to correct for the number of RBCs in a traumatic lumbar puncture is to allow 1 WBC for every 700 RBCs/L. Given this conversion, there are still too many WBCs in this sample, indicating a mild pleocytosis that is approximately one‐half neutrophilic and one‐half lymphocytic. This profile is nonspecific and can occur with a variety of conditions including stroke, seizure, inflammatory disorders, and infections, including viruses such as West Nile virus.
While coverage with acyclovir and broad‐spectrum antibacterials is appropriate, it should be noted that piperacillin‐tazobactam has poor CSF penetration and therefore is not a good choice for empiric coverage of CNS infections.
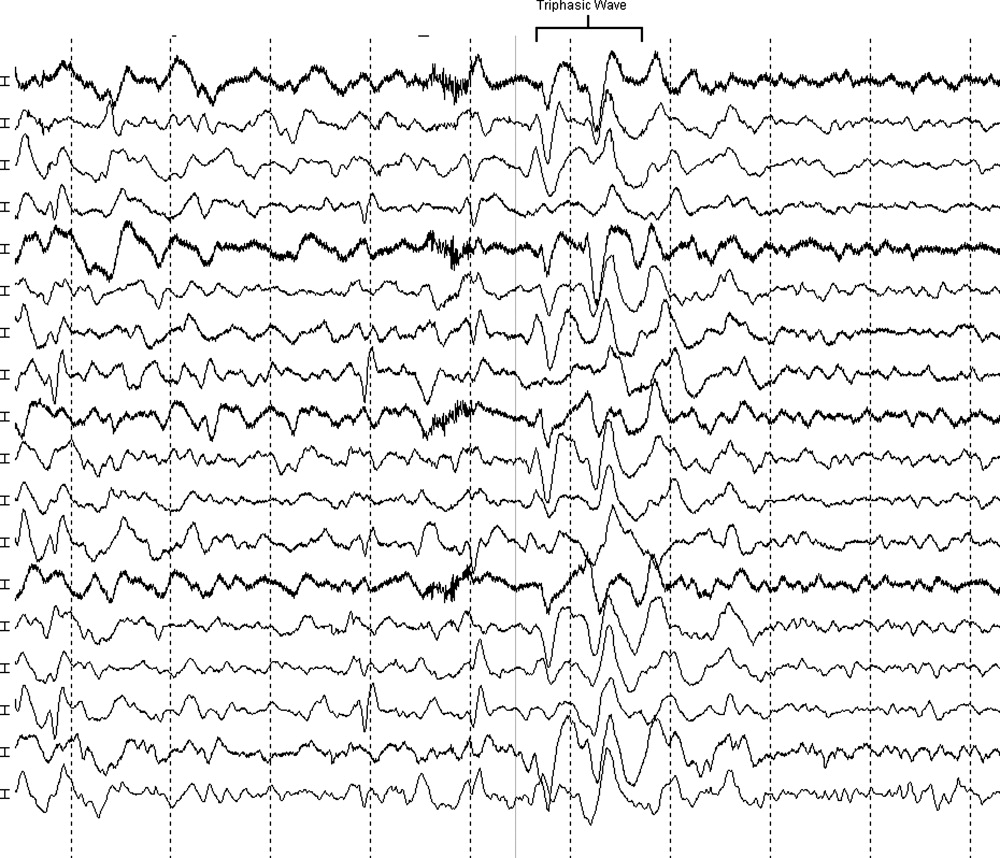
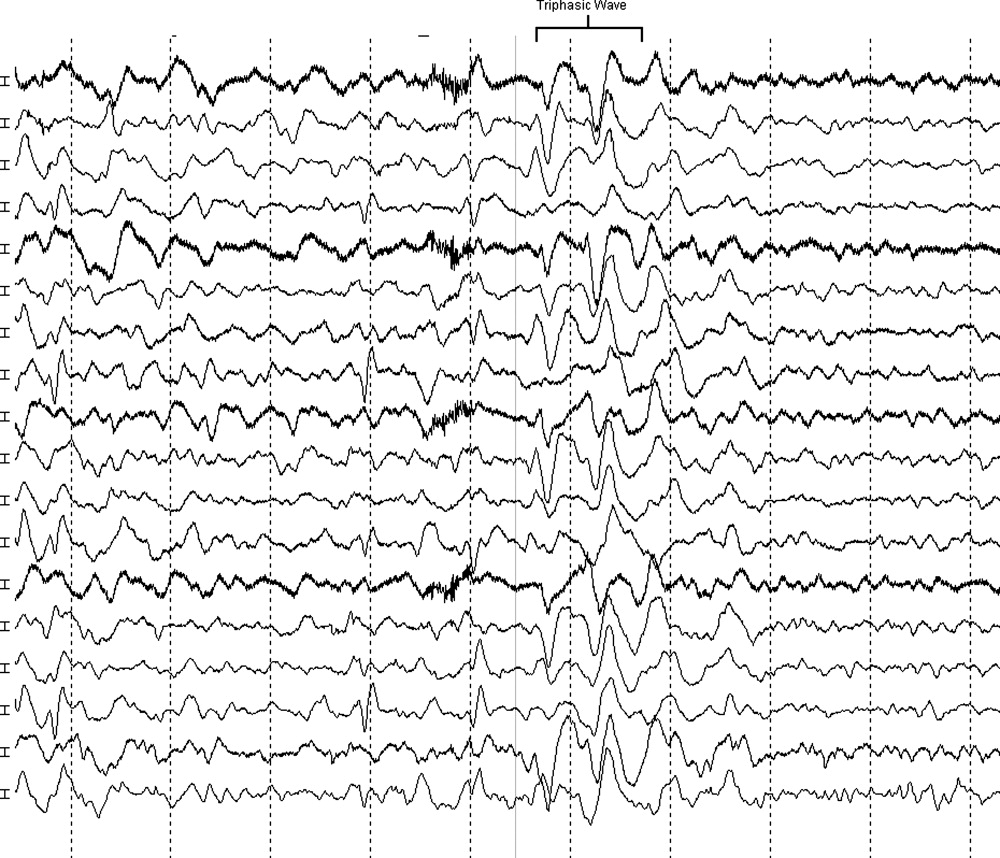
The neurologist's examination additionally noted multifocal myoclonus with noxious stimuli, most prominent in the face and toes. An urgent EEG demonstrated continuous, slow, generalized triphasic wave activity (Figures 1 and 2); no epileptiform discharges were seen. The erythrocyte sedimentation rate (ESR) was 66 mm/hour (normal, 0‐30), and tests for antinuclear antibodies, serum levetiracetam level, and thyroid function studies were ordered.
Stimulus‐evoked multifocal myoclonus is a general marker of encephalopathy found in many conditions, including hepatic and renal failure, drug intoxication (eg, opiates), neurodegenerative disorders (eg, Creutzfeldt‐Jakob disease [CJD]), and postanoxic injury, the latter of which is termed the Lance‐Adams syndrome.
Triphasic waves on EEG, while commonly associated with hepatic encephalopathy, have a similarly broad differential diagnosis, although in a comatose patient, they must first and foremost be distinguished from the repetitive discharges characteristic of nonconvulsive status epilepticus. In addition to hepatic and renal failure, triphasic waves have also been described in medication toxicity (especially with anticonvulsants, lithium, and cephalosporins), CNS infections (including Lyme disease and West Nile virus), strokes involving the bilateral thalami (usually from deep venous thrombosis), inflammatory disorders (such as Hashimoto's encephalopathy [HE]), and neurodegenerative diseases. It is important to remember that a single EEG does not exclude the possibility of an episodic ictal disorder and longer‐term monitoring would be required to definitively exclude seizures.
At this point, although the myoclonus and triphasic waves most commonly would indicate a toxic/metabolic process, the elevated ESR and CSF pleocytosis argue for an inflammatory or infectious condition. An MRI remains the next most useful test to guide further workup because many such conditions have distinct signatures on MRI.
The following day, she was noted to have periods of alertnessopening her eyes and following some commandsbut at other times she was difficult to arouse or obtunded. Tremulous movements and sporadic myoclonic jerks continued but no focal neurologic signs were found. Although there was increased muscle tone throughout, she was intermittently seen moving her limbs spontaneously, but not to command. No new findings were appreciated on routine laboratory tests. Antinuclear antibody testing was negative. Serum levetiracetam level was 23.5 g/mL (reference range, 545). Serum thyroid‐stimulating hormone was less than 0.005 U/mL, but free T3 was 3.5 pg/mL (normal, 1.8‐4.6) and free T4 was 2.0 ng/dL (normal, 0.71.8). An MRI of the brain was compromised by motion artifact but no significant abnormalities were appreciated.
At this point, a family member in another state disclosed that the patient had also been hospitalized 2 months previously while visiting him. Her chief complaint had been shortness of breath. The records were obtained; a cardiac catheterization had revealed nonobstructive coronary disease, and medical management was recommended. The notes mentioned that during the hospitalization she developed altered mental status with disorientation and shaking. CT and MRI of the brain had been unremarkable. The confusion was not explained, but she was discharged in good condition, alert and fully‐oriented.
The additional history confirms a relapsing encephalopathy, now with at least 3 occurrences. The most common etiologies in the face of a normal MRI and basic labs would be recurrent intoxication or exposures, but the inflammatory CSF profile and elevated ESR are not consistent with this. A variety of inflammatory disorders can present with recurrent encephalopathy, including demyelinating diseases and neurosarcoidosis. Some systemic rheumatologic conditions, such as systemic lupus erythematosus, can present with relapsing neurologic symptoms due to seizures, vasculitis, or cerebritis. Vasculitis would fit this picture as well, except for the normal findings on 2 MRIs. In a patient with such dramatic symptoms of neurologic dysfunction, one would expect to see changes on the MRI of cerebral inflammation with probable ischemia.
Therefore, given the CSF, ESR, clinical course, and unrevealing MRI and EEG, the most likely group of disorders responsible would be the nonvasculitic autoimmune meningoencephalitides, which present with recurrent encephalopathy and feature spontaneous remissions and/or often‐dramatic responses to corticosteroids. Key disorders in this category include Sjogren's disease, lupus, and steroid responsive encephalopathy associated with autoimmune thyroiditis (Hashimoto's encephalopathy). The latter condition is the most common of the group and is suggested by the abnormal thyroid‐stimulating hormone testing, although it may occur in the setting of normal thyroid function. The diagnosis can be confirmed with thyroperoxidase and thyroglobulin antibody testing.
Three days into the hospitalization, her mental status had gradually improved such that she was more consistently awake and oriented to person and place, and she was transferred to a regular nursing unit. Final results from the CSF and blood cultures were negative, as was PCR for HSV. The antimicrobials were discontinued. Routine serum chemistries continued to be unremarkable. Additional studies recommended by the neurologist demonstrated an antithyroperoxidase antibody concentration of 587.1 IU/mL (normal, <5), and antithyroglobulin antibody level of 52.2 IU/mL (normal, <10).
These results confirm the diagnosis of HE which, in addition to its presentation as a recurrent illness, is an important treatable cause of dementia and should be considered in young patients, those with autoimmune and thyroid disorders, and those whose dementia is rapidly progressive. Most cases are thought to be steroid‐responsive, but some studies have defined the disorder based on this responsiveness, resulting in some nonresponders likely being overlooked.
A trial of corticosteroids should be considered if the patient does not quickly return to baseline given the potential morbidities associated with prolonged altered mental status to this degree. Whether initiation of chronic immunosuppression could prevent these attacks in the future is unclear from the literature but should be considered given the recurrent, dramatic presentation in this patient.
A diagnosis of HE was made, and she was prescribed corticosteroids. Twenty‐four hours later, she was alert and fully‐oriented. She was discharged to home on prednisone and seen in follow‐up in neurology clinic 1 month later. She had had no further episodes of confusion or stupor, but because of steroid‐induced hyperglycemia, her corticosteroids were decreased and mycophenolate mofetil added for chronic immunosuppression. Four months after discharge she was neurologically stable but continued to struggle with the adverse effects of chronic corticosteroid treatment.
COMMENTARY
HE is an uncommon condition that can present with a rapidly progressive decline and should be considered in patients who present with recurrent mental status change in the setting of normal imaging studies and routine laboratory results. The entity was initially described by Lord William Russell Brain in 1966, and in the most recent terminology is known as steroid‐responsive encephalopathy associated with autoimmune thyroiditis (SREAT).1 It is characterized by an acute or subacute encephalopathy associated with thyroid autoimmunity. Patients typically present with fluctuating symptoms, episodes of confusion, alterations of consciousness, and rapid cognitive decline.2 Common features include myoclonus, tremor, ataxia, speech disturbance, stroke‐like episodes, increased muscle tone, neuropsychiatric manifestations, and seizures, that in some cases may progress to status epilepticus.3, 4
Although serum antithyroglobulin and antithyroperoxidase antibodies are elevated in HE, their presence is thought to be an epiphenomenon of the condition rather than the direct cause. Supporting this are the facts that the incidence of encephalopathy is not increased in patients with established autoimmune thyroiditis, and the presence of antithyroid antibodies ranges from 5% to 20% in the general population.2, 5 There is also no evidence that thyroid antibodies directly react with brain tissue, and the levels of these antibodies do not correlate with either neurologic manifestations or clinical improvement.2, 4, 5 As HE has been reported in patients with euthyroidism, hypothyroidism, and hyperthyroidism (with hypothyroidismeither subclinical or activemost common), it is also unlikely that the level of thyroid hormones play a role in the etiology of this disease.2, 4, 6
The etiology and pathogenesis of HE are unclear, although an immune‐mediated process is generally implicated, either from an inflammatory vasculitis or as a form of acute disseminated encephalomyelitis.7‐9 Global hypoperfusion on single‐photon emission computed tomography (SPECT) studies has also been reported.10, 11 Patients with HE may have nonspecific evidence of inflammation, including an elevated ESR, CRP, and CSF protein.12 Other laboratory abnormalities may include a mild elevation of liver aminotransferase levels; renal impairment has also been reported in a few cases of HE in the form of glomerulonephritis, and may be related to deposition of immune complexes containing thyroglobulin antigen.6, 12‐14 MRI of the brain is normal or nonspecific in most cases, and the EEG most commonly shows diffuse slowing.
The differential for a rapidly progressive cognitive decline includes CJD, CNS vasculitis, paraneoplastic syndromes, and autoimmune and subacute infectious encephalopathies. In patients with CJD, T2‐weighted imaging may show hyperintense signals in the basal ganglia, while diffusion‐weighted sequences may reveal changes in the cortical ribbon and bilateral thalami.15 In CNS vasculitis, the imaging findings are variable and range from discrete areas of vascular infarcts to hemorrhagic lesions.16 In paraneoplastic and autoimmune encephalopathies (excluding HE), MRI often shows nonenhancing signal intensity changes in the mesial temporal lobes.12 This patient had repeatedly normal MRI studies of the brain, which in combination with the history of tremor, myoclonus, seizures, and interval return to baseline status, helped point to the diagnosis of HE.
Different approaches to treatment of HE have been recommended. As the acronym SREAT suggests, patients typically respond dramatically to high‐dose steroid therapy. Although a number of patients also improve spontaneously, up to 60% of patients experience a relapsing course and require chronic immunosuppressive agents for maintenance therapy, including long‐term steroids and azathioprine.2, 17 Treatment with plasma exchange and intravenous immune globulin have also been reported, but with mixed results.18, 19 Due to her history of multiple relapses, the patient was placed on mycophenolate mofetil for additional maintenance immunosuppression, as her corticosteroid dose was reduced due to adverse effects.
Acute mental status change is a potentially emergent situation that must be evaluated with careful history and studies to exclude life‐threatening metabolic, infectious, and vascular conditions. This patient presented similarly on 2 prior occasions, and each time her physician team evaluated what appeared to be a new onset of altered consciousness, reaching a plausible but ultimately incorrect diagnosis. The patient's third presentation was finally the charm, as her physicians learned of the repeated history of a confusional state, and in particular the return to baseline status, allowing them to create a differential that focused on etiologies of relapsing encephalopathy and make the correct diagnosis.
Key Points
-
Recurrent acute or subacute cognitive deterioration invokes a differential diagnosis of toxic/metabolic disorders and unusual inflammatory conditions.
-
The nonvasculitic autoimmune encephalopathies are a group of uncommon conditions characterized by nonspecific findings of inflammation and generally unremarkable CNS imaging studies.
-
HE, or SREAT, is the most common of these conditions, and is notable for mental status changes, various findings of increased muscular tone, thyroid autoimmunity, and generally a dramatic response to corticosteroids.
- , , .Hashimoto's disease and encephalopathy.Lancet.1966;2:512–514.
- , , .Hashimoto encephalopathy: syndrome or myth?Arch Neurol.2003;60:164–171.
- , , .Pisani F. Recurrent status epilepticus as the main feature of Hashimoto's encephalopathy.Epilepsy Behav.2006;8:328–330.
- , , , et al.Steroid‐responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol.2006;63:197–202.
- , , , , .Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment.J Neurol.1996;243:585–593.
- , , , , .Hashimoto's encephalopathy: a steroid‐responsive disorder associated with high anti‐thyroid antibody titers‐report of 5 cases.Neurology.1991;41:228–233.
- , , , , .Hashimoto encephalopathy: a brainstem vasculitis?Neurology.2000;54:769–770.
- , , , , .Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): A reversible form of encephalopathy.Neurology.1999;53:1579–1581.
- , , , .Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus.Neurology.2003;61:1124–1126.
- , , .Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism.Neurology.1997;49:623–626.
- , , , , .Hashimoto's encephalopathy: clinical, SPECT and neurophysiologic data.QJM.2003;96:455–457.
- , , .Autoimmune Encephalopathies.The Neurologist.2007;13:140–147.
- , , , , , .Thyroid antigen‐antibody nephritis.Clin Immunol Immunopathol1976;6:341–346.
- , , .Immune complex glomerulonephritis mediated by thyroid antigens.Arch Pathol Lab Med1978;102:530–533.
- , , , et al.Diffusion‐weighted MR imaging of early‐stage Creutzfeldt‐Jakob disease: typical and atypical manifestations.Radiographics.2006;26:S191–S204.
- , , , , .CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.AJNR Am J Neuroradiol.1999;20:75–85.
- , .Long‐Term Treatment of Hashimoto's Encephalopathy.J Neuropsychiatry Clin Neurosci.2006;18:14–20.
- , .Hashimoto's encephalopathy: steroid resistance and response to intravenouc immunoglobulins.J Neurol Neurosurg Psychiatry.2005;76:455–456.
- , .Hashimoto's encephalopathy responding to plasmapheresis.J Neurol Neurosurg Psychiatry.2001;70:132.
A 58‐year old woman was brought to the emergency department with confusion. Her husband stated that for several hours she had been drifting in and out at home, and that he had to shout to get her attention. He described no seizure activity, weakness, incontinence, or difficulty speaking, and had noted no complaints of headache, fevers, chest pain, shortness of breath, or gastrointestinal complaints.
Altered mental status in a middle‐aged woman can result from a diverse set of etiologies. A key distinction in the neurological examination will be to assure that the complaint of confusion is accurate as opposed to aphasia; the former is usually indicative of diffuse cerebral dysfunction while the latter suggests a focal lesion in the dominant hemisphere.
The acuity of the change in mental status is important, as are the fluctuations described by the husband. Unwitnessed or nonconvulsive seizure activity can present this way. Toxic/metabolic etiologies, infectious and inflammatory disorders of the central nervous system (CNS), and vascular diseases are also important considerations. Although stroke does not typically present with global encephalopathy, intermittent large vessel occlusion, especially in the posterior circulation, can disrupt cognition in this manner. Following a physical examination, initial workup should focus on toxic/metabolic etiologies, followed rapidly by head imaging if no cause is identified.
Her past medical history was notable for type 2 diabetes mellitus, coronary artery disease, hyperlipidemia, and an unspecified seizure disorder, which according to her husband was diagnosed during a recent hospitalization for a similar presentation. She also had a remote history of venous thromboembolism and antithrombin‐III deficiency. She was unemployed, lived with her husband, and spent most of her time at home. She never smoked, and rarely drank alcohol. Her family history was unobtainable, and her husband denied that she used any illicit drugs. Her medications included pioglitazone, aspirin, simvastatin, pregabalin, ferrous sulfate, levetiracetam, warfarin, and magnesium oxide, and she was allergic to sulfa.
While the differential diagnosis remains broad, 3 elements of the history are potentially relevant. The history of epilepsy based on a similar prior presentation increases the likelihood that the current spell is ictal in nature; examination of previous records would be important in order to document whether these spells have indeed been proven to be epileptic, as many conditions can mimic seizures. Given the history of venous thromboembolism and hypercoagulability, one must consider cerebral venous sinus thrombosis, which can present with global neurologic dysfunction and seizures. Prompt identification, usually via computed tomography (CT) or magnetic resonance angiography, is vital, because anticoagulation can mitigate this potentially life‐threatening illness. Finally, although many medications can cause encephalopathy in overdose, levetiracetam has well‐described cognitive side effects even at usual doses, including encephalopathy, irritability, and depression.
The records from that recent hospitalization remarked that she had presented confused and stuporous. Her potassium had been 2.7 mmol/L, international normalized ration (INR) 3.4, and hemoglobin 8 g/dL; other routine laboratory studies were normal. CT and magnetic resonance imaging (MRI) of the brain had been negative, and electroencephalogram (EEG) reportedly was performed but specific results were unknown. She was discharged alert and oriented 1 week prior to the current presentation on the above medications, including levetiracetam for this newly‐diagnosed seizure disorder.
Previous records confirm that the current presentation is that of a relapsing acute alteration in mental status. Regardless of the EEG findings or response to antiepileptic medications, a seizure disorder should remain a primary consideration, although relapsing inflammatory, toxic/metabolic conditions, and, rarely, vascular disorders can also present in this manner.
The neurologic manifestations of hypokalemia are usually peripheral in nature, including periodic paralysis; confusion accompanying hypokalemia is usually not a result of the low potassium itself but rather due to an underlying toxic or endocrinologic cause. Various causes of anemia can lead to mental status changes; the mean corpuscular volume (MCV) will be particularly helpful given known associations between megaloblastic anemia and confusional states.
On examination, she appeared to be in good health and in no distress. She was afebrile. Her blood pressure was 93/57, pulse 90 beats per minute, respiratory rate 16 per minute, and room air oxygen saturation 100%. She was oriented to her surroundings, but slow in her responses to questioning. There were no cranial nerve, motor, or sensory deficits, or abnormal reflexes or movements. Examination of the head, skin, chest, cardiovascular system, abdomen, and extremities was normal. Serum sodium was 136 mmol/L, creatinine 1.2 mg/dL, calcium 9.3 mg/dL, and glucose 81 mg/dL; other routine blood chemistries were normal. Her white blood cell (WBC) count was 7100/L, hemoglobin 9.2 g/dL with normal MCV, and platelet count 275,000/L. INR was 3.4, and liver function tests were normal. CT of the brain demonstrated no evidence of acute pathology.
Given that her laboratory results (aside from the hemoglobin) and CT were essentially normal, the most common etiology of a recurrent encephalopathy would be a toxic exposure including drugs, alcohol, and environmental toxins or poisons. A comprehensive serum drug screen, including heavy metals, could follow a basic urinary screen for drugs of abuse; specific etiologies may be suggested by patterns of injury seen on MRI such as those seen with carbon monoxide or methanol exposure. Other recurrent metabolic processes include the porphyrias and relapsing inflammatory disorders, which could be entertained if further diagnostics are unrevealing.
An EEG is warranted at this point and is a test that is underutilized in the workup of altered mental status. Patients who have a spell and do not quickly awaken should be considered to be in nonconvulsive status epilepticus until proven otherwise. This can be easily identified on the EEG and is an important entity to recognize quickly. Additional findings on EEG may suggest focal cerebral dysfunction (such as that following a seizure or acute unilateral injury), diffuse encephalopathy (eg, triphasic waves), or fairly specific diagnoses (eg, periodic lateralized epileptiform discharges from the temporal lobes in suspected herpes simplex meningoencephalitis). While the CT of the brain is a reasonable initial screen, MRI is more sensitive for structural disease and should be obtained if no etiology is rapidly identified.
Finally, acute infectious etiologies such as abscess, encephalitis, or meningoencephalitis need to be excluded via lumbar puncture. Spinal fluid examination also can be helpful in the consideration of inflammatory and autoimmune disorders.
Over the next several hours, while still in the emergency department, she became increasingly obtunded, to the point that she was unresponsive to all stimuli. No seizure activity was witnessed, her vital signs were unchanged, and no medications had been administered. She was urgently transferred to a tertiary care center, where, at the time of arrival, she was obtunded and nonverbal, and opened her eyes only to noxious stimuli. She would withdraw all 4 extremities in response to pain. Pupils were 2 mm and symmetrically reactive. Corneal reflexes were normal, and her gag reflex was diminished. Motor tone was decreased in all 4 extremities. No fasciculations were noted. Deep tendon reflexes were present but symmetrically diminished throughout, and Babinski testing demonstrated a withdrawal response bilaterally.
Coma is a state of profound unconsciousness where the patient is unarousable and unaware of her surroundings. Coma can result either from bihemispheric dysfunction or diffuse injury to the reticular activating system in the brainstem, and the physical examination should focus on distinguishing between these 2 sites. Because the nuclei of cranial nerves III through XII (excepting XI) reside in the brainstem, the coma examination emphasizes testing the cranial nerves; although all cranial nerves are not tested in this patient, the ones that are appear to be normal, making bihemispheric dysfunction most likely. Bihemispheric coma most commonly results from diffuse toxic or metabolic etiologies such as intoxication or hepatic encephalopathy, but it can also be caused by bilateral structural lesions (including the bilateral thalami) or ongoing seizure activity.
Although an EEG remains the key test in this patient given her past history and an MRI would prove extremely useful, her deterioration warrants a workup for CNS infection. Since the head CT was negative, it would be prudent to proceed with urgent lumbar puncture (although it should never be performed in a patient with significant coagulopathy due to risks of hemorrhage leading to spinal cord injury). She should be covered empirically with broad spectrum meningeal‐dose antibiotics, including acyclovir, until the results of the spinal fluid examination are known, given that bacterial meningitis and herpes meningoencephalitis carry a high morbidity and mortality if not treated promptly.
Routine blood tests were similar to her labs at the referring emergency room. Ammonia level was 10 mol/L. Urine toxicology screen was negative, and blood tests for ethanol, salicylates, lithium, and acetaminophen were negative. Chest X‐ray and urinalysis were normal, and electrocardiogram was notable only for a sinus tachycardia. Cultures of the blood were obtained and the patient was admitted to the intensive care unit.
Levetiracetam, vancomycin, piperacillin‐tazobactam, and acyclovir were initiated. A lumbar puncture was performed without reversing the anticoagulation, and the procedure was traumatic. The cerebrospinal fluid was bloody, with a clear supernatant. Cell count demonstrated a red blood cell (RBC) count of 1250/L and a WBC count of 9/L, with a WBC differential of 42% neutrophils, 48% lymphocytes, and 8% monocytes. The cerebrospinal fluid (CSF) glucose was 62 mg/dL (with a serum glucose of 74 mg/dL) and protein 41 mg/dL. The CSF Gram stain demonstrated no organisms, and fluid was sent for routine culture and polymerase chain reaction (PCR) to detect herpes simplex virus (HSV). A neurology consultation was urgently requested.
As mentioned, it would have been more appropriate to reverse the patient's anticoagulation prior to lumbar puncture. The absence of xanthochromia suggests that the RBCs seen in the sample were introduced at the time of the lumbar puncture, arguing against a hemorrhagic disorder of the CNS (occasionally seen with herpes simplex encephalitis) or spinal fluid (eg, subarachnoid hemorrhage).
A reasonable rule of thumb to correct for the number of RBCs in a traumatic lumbar puncture is to allow 1 WBC for every 700 RBCs/L. Given this conversion, there are still too many WBCs in this sample, indicating a mild pleocytosis that is approximately one‐half neutrophilic and one‐half lymphocytic. This profile is nonspecific and can occur with a variety of conditions including stroke, seizure, inflammatory disorders, and infections, including viruses such as West Nile virus.
While coverage with acyclovir and broad‐spectrum antibacterials is appropriate, it should be noted that piperacillin‐tazobactam has poor CSF penetration and therefore is not a good choice for empiric coverage of CNS infections.
The neurologist's examination additionally noted multifocal myoclonus with noxious stimuli, most prominent in the face and toes. An urgent EEG demonstrated continuous, slow, generalized triphasic wave activity (Figures 1 and 2); no epileptiform discharges were seen. The erythrocyte sedimentation rate (ESR) was 66 mm/hour (normal, 0‐30), and tests for antinuclear antibodies, serum levetiracetam level, and thyroid function studies were ordered.
Stimulus‐evoked multifocal myoclonus is a general marker of encephalopathy found in many conditions, including hepatic and renal failure, drug intoxication (eg, opiates), neurodegenerative disorders (eg, Creutzfeldt‐Jakob disease [CJD]), and postanoxic injury, the latter of which is termed the Lance‐Adams syndrome.
Triphasic waves on EEG, while commonly associated with hepatic encephalopathy, have a similarly broad differential diagnosis, although in a comatose patient, they must first and foremost be distinguished from the repetitive discharges characteristic of nonconvulsive status epilepticus. In addition to hepatic and renal failure, triphasic waves have also been described in medication toxicity (especially with anticonvulsants, lithium, and cephalosporins), CNS infections (including Lyme disease and West Nile virus), strokes involving the bilateral thalami (usually from deep venous thrombosis), inflammatory disorders (such as Hashimoto's encephalopathy [HE]), and neurodegenerative diseases. It is important to remember that a single EEG does not exclude the possibility of an episodic ictal disorder and longer‐term monitoring would be required to definitively exclude seizures.
At this point, although the myoclonus and triphasic waves most commonly would indicate a toxic/metabolic process, the elevated ESR and CSF pleocytosis argue for an inflammatory or infectious condition. An MRI remains the next most useful test to guide further workup because many such conditions have distinct signatures on MRI.
The following day, she was noted to have periods of alertnessopening her eyes and following some commandsbut at other times she was difficult to arouse or obtunded. Tremulous movements and sporadic myoclonic jerks continued but no focal neurologic signs were found. Although there was increased muscle tone throughout, she was intermittently seen moving her limbs spontaneously, but not to command. No new findings were appreciated on routine laboratory tests. Antinuclear antibody testing was negative. Serum levetiracetam level was 23.5 g/mL (reference range, 545). Serum thyroid‐stimulating hormone was less than 0.005 U/mL, but free T3 was 3.5 pg/mL (normal, 1.8‐4.6) and free T4 was 2.0 ng/dL (normal, 0.71.8). An MRI of the brain was compromised by motion artifact but no significant abnormalities were appreciated.
At this point, a family member in another state disclosed that the patient had also been hospitalized 2 months previously while visiting him. Her chief complaint had been shortness of breath. The records were obtained; a cardiac catheterization had revealed nonobstructive coronary disease, and medical management was recommended. The notes mentioned that during the hospitalization she developed altered mental status with disorientation and shaking. CT and MRI of the brain had been unremarkable. The confusion was not explained, but she was discharged in good condition, alert and fully‐oriented.
The additional history confirms a relapsing encephalopathy, now with at least 3 occurrences. The most common etiologies in the face of a normal MRI and basic labs would be recurrent intoxication or exposures, but the inflammatory CSF profile and elevated ESR are not consistent with this. A variety of inflammatory disorders can present with recurrent encephalopathy, including demyelinating diseases and neurosarcoidosis. Some systemic rheumatologic conditions, such as systemic lupus erythematosus, can present with relapsing neurologic symptoms due to seizures, vasculitis, or cerebritis. Vasculitis would fit this picture as well, except for the normal findings on 2 MRIs. In a patient with such dramatic symptoms of neurologic dysfunction, one would expect to see changes on the MRI of cerebral inflammation with probable ischemia.
Therefore, given the CSF, ESR, clinical course, and unrevealing MRI and EEG, the most likely group of disorders responsible would be the nonvasculitic autoimmune meningoencephalitides, which present with recurrent encephalopathy and feature spontaneous remissions and/or often‐dramatic responses to corticosteroids. Key disorders in this category include Sjogren's disease, lupus, and steroid responsive encephalopathy associated with autoimmune thyroiditis (Hashimoto's encephalopathy). The latter condition is the most common of the group and is suggested by the abnormal thyroid‐stimulating hormone testing, although it may occur in the setting of normal thyroid function. The diagnosis can be confirmed with thyroperoxidase and thyroglobulin antibody testing.
Three days into the hospitalization, her mental status had gradually improved such that she was more consistently awake and oriented to person and place, and she was transferred to a regular nursing unit. Final results from the CSF and blood cultures were negative, as was PCR for HSV. The antimicrobials were discontinued. Routine serum chemistries continued to be unremarkable. Additional studies recommended by the neurologist demonstrated an antithyroperoxidase antibody concentration of 587.1 IU/mL (normal, <5), and antithyroglobulin antibody level of 52.2 IU/mL (normal, <10).
These results confirm the diagnosis of HE which, in addition to its presentation as a recurrent illness, is an important treatable cause of dementia and should be considered in young patients, those with autoimmune and thyroid disorders, and those whose dementia is rapidly progressive. Most cases are thought to be steroid‐responsive, but some studies have defined the disorder based on this responsiveness, resulting in some nonresponders likely being overlooked.
A trial of corticosteroids should be considered if the patient does not quickly return to baseline given the potential morbidities associated with prolonged altered mental status to this degree. Whether initiation of chronic immunosuppression could prevent these attacks in the future is unclear from the literature but should be considered given the recurrent, dramatic presentation in this patient.
A diagnosis of HE was made, and she was prescribed corticosteroids. Twenty‐four hours later, she was alert and fully‐oriented. She was discharged to home on prednisone and seen in follow‐up in neurology clinic 1 month later. She had had no further episodes of confusion or stupor, but because of steroid‐induced hyperglycemia, her corticosteroids were decreased and mycophenolate mofetil added for chronic immunosuppression. Four months after discharge she was neurologically stable but continued to struggle with the adverse effects of chronic corticosteroid treatment.
COMMENTARY
HE is an uncommon condition that can present with a rapidly progressive decline and should be considered in patients who present with recurrent mental status change in the setting of normal imaging studies and routine laboratory results. The entity was initially described by Lord William Russell Brain in 1966, and in the most recent terminology is known as steroid‐responsive encephalopathy associated with autoimmune thyroiditis (SREAT).1 It is characterized by an acute or subacute encephalopathy associated with thyroid autoimmunity. Patients typically present with fluctuating symptoms, episodes of confusion, alterations of consciousness, and rapid cognitive decline.2 Common features include myoclonus, tremor, ataxia, speech disturbance, stroke‐like episodes, increased muscle tone, neuropsychiatric manifestations, and seizures, that in some cases may progress to status epilepticus.3, 4
Although serum antithyroglobulin and antithyroperoxidase antibodies are elevated in HE, their presence is thought to be an epiphenomenon of the condition rather than the direct cause. Supporting this are the facts that the incidence of encephalopathy is not increased in patients with established autoimmune thyroiditis, and the presence of antithyroid antibodies ranges from 5% to 20% in the general population.2, 5 There is also no evidence that thyroid antibodies directly react with brain tissue, and the levels of these antibodies do not correlate with either neurologic manifestations or clinical improvement.2, 4, 5 As HE has been reported in patients with euthyroidism, hypothyroidism, and hyperthyroidism (with hypothyroidismeither subclinical or activemost common), it is also unlikely that the level of thyroid hormones play a role in the etiology of this disease.2, 4, 6
The etiology and pathogenesis of HE are unclear, although an immune‐mediated process is generally implicated, either from an inflammatory vasculitis or as a form of acute disseminated encephalomyelitis.7‐9 Global hypoperfusion on single‐photon emission computed tomography (SPECT) studies has also been reported.10, 11 Patients with HE may have nonspecific evidence of inflammation, including an elevated ESR, CRP, and CSF protein.12 Other laboratory abnormalities may include a mild elevation of liver aminotransferase levels; renal impairment has also been reported in a few cases of HE in the form of glomerulonephritis, and may be related to deposition of immune complexes containing thyroglobulin antigen.6, 12‐14 MRI of the brain is normal or nonspecific in most cases, and the EEG most commonly shows diffuse slowing.
The differential for a rapidly progressive cognitive decline includes CJD, CNS vasculitis, paraneoplastic syndromes, and autoimmune and subacute infectious encephalopathies. In patients with CJD, T2‐weighted imaging may show hyperintense signals in the basal ganglia, while diffusion‐weighted sequences may reveal changes in the cortical ribbon and bilateral thalami.15 In CNS vasculitis, the imaging findings are variable and range from discrete areas of vascular infarcts to hemorrhagic lesions.16 In paraneoplastic and autoimmune encephalopathies (excluding HE), MRI often shows nonenhancing signal intensity changes in the mesial temporal lobes.12 This patient had repeatedly normal MRI studies of the brain, which in combination with the history of tremor, myoclonus, seizures, and interval return to baseline status, helped point to the diagnosis of HE.
Different approaches to treatment of HE have been recommended. As the acronym SREAT suggests, patients typically respond dramatically to high‐dose steroid therapy. Although a number of patients also improve spontaneously, up to 60% of patients experience a relapsing course and require chronic immunosuppressive agents for maintenance therapy, including long‐term steroids and azathioprine.2, 17 Treatment with plasma exchange and intravenous immune globulin have also been reported, but with mixed results.18, 19 Due to her history of multiple relapses, the patient was placed on mycophenolate mofetil for additional maintenance immunosuppression, as her corticosteroid dose was reduced due to adverse effects.
Acute mental status change is a potentially emergent situation that must be evaluated with careful history and studies to exclude life‐threatening metabolic, infectious, and vascular conditions. This patient presented similarly on 2 prior occasions, and each time her physician team evaluated what appeared to be a new onset of altered consciousness, reaching a plausible but ultimately incorrect diagnosis. The patient's third presentation was finally the charm, as her physicians learned of the repeated history of a confusional state, and in particular the return to baseline status, allowing them to create a differential that focused on etiologies of relapsing encephalopathy and make the correct diagnosis.
Key Points
-
Recurrent acute or subacute cognitive deterioration invokes a differential diagnosis of toxic/metabolic disorders and unusual inflammatory conditions.
-
The nonvasculitic autoimmune encephalopathies are a group of uncommon conditions characterized by nonspecific findings of inflammation and generally unremarkable CNS imaging studies.
-
HE, or SREAT, is the most common of these conditions, and is notable for mental status changes, various findings of increased muscular tone, thyroid autoimmunity, and generally a dramatic response to corticosteroids.
A 58‐year old woman was brought to the emergency department with confusion. Her husband stated that for several hours she had been drifting in and out at home, and that he had to shout to get her attention. He described no seizure activity, weakness, incontinence, or difficulty speaking, and had noted no complaints of headache, fevers, chest pain, shortness of breath, or gastrointestinal complaints.
Altered mental status in a middle‐aged woman can result from a diverse set of etiologies. A key distinction in the neurological examination will be to assure that the complaint of confusion is accurate as opposed to aphasia; the former is usually indicative of diffuse cerebral dysfunction while the latter suggests a focal lesion in the dominant hemisphere.
The acuity of the change in mental status is important, as are the fluctuations described by the husband. Unwitnessed or nonconvulsive seizure activity can present this way. Toxic/metabolic etiologies, infectious and inflammatory disorders of the central nervous system (CNS), and vascular diseases are also important considerations. Although stroke does not typically present with global encephalopathy, intermittent large vessel occlusion, especially in the posterior circulation, can disrupt cognition in this manner. Following a physical examination, initial workup should focus on toxic/metabolic etiologies, followed rapidly by head imaging if no cause is identified.
Her past medical history was notable for type 2 diabetes mellitus, coronary artery disease, hyperlipidemia, and an unspecified seizure disorder, which according to her husband was diagnosed during a recent hospitalization for a similar presentation. She also had a remote history of venous thromboembolism and antithrombin‐III deficiency. She was unemployed, lived with her husband, and spent most of her time at home. She never smoked, and rarely drank alcohol. Her family history was unobtainable, and her husband denied that she used any illicit drugs. Her medications included pioglitazone, aspirin, simvastatin, pregabalin, ferrous sulfate, levetiracetam, warfarin, and magnesium oxide, and she was allergic to sulfa.
While the differential diagnosis remains broad, 3 elements of the history are potentially relevant. The history of epilepsy based on a similar prior presentation increases the likelihood that the current spell is ictal in nature; examination of previous records would be important in order to document whether these spells have indeed been proven to be epileptic, as many conditions can mimic seizures. Given the history of venous thromboembolism and hypercoagulability, one must consider cerebral venous sinus thrombosis, which can present with global neurologic dysfunction and seizures. Prompt identification, usually via computed tomography (CT) or magnetic resonance angiography, is vital, because anticoagulation can mitigate this potentially life‐threatening illness. Finally, although many medications can cause encephalopathy in overdose, levetiracetam has well‐described cognitive side effects even at usual doses, including encephalopathy, irritability, and depression.
The records from that recent hospitalization remarked that she had presented confused and stuporous. Her potassium had been 2.7 mmol/L, international normalized ration (INR) 3.4, and hemoglobin 8 g/dL; other routine laboratory studies were normal. CT and magnetic resonance imaging (MRI) of the brain had been negative, and electroencephalogram (EEG) reportedly was performed but specific results were unknown. She was discharged alert and oriented 1 week prior to the current presentation on the above medications, including levetiracetam for this newly‐diagnosed seizure disorder.
Previous records confirm that the current presentation is that of a relapsing acute alteration in mental status. Regardless of the EEG findings or response to antiepileptic medications, a seizure disorder should remain a primary consideration, although relapsing inflammatory, toxic/metabolic conditions, and, rarely, vascular disorders can also present in this manner.
The neurologic manifestations of hypokalemia are usually peripheral in nature, including periodic paralysis; confusion accompanying hypokalemia is usually not a result of the low potassium itself but rather due to an underlying toxic or endocrinologic cause. Various causes of anemia can lead to mental status changes; the mean corpuscular volume (MCV) will be particularly helpful given known associations between megaloblastic anemia and confusional states.
On examination, she appeared to be in good health and in no distress. She was afebrile. Her blood pressure was 93/57, pulse 90 beats per minute, respiratory rate 16 per minute, and room air oxygen saturation 100%. She was oriented to her surroundings, but slow in her responses to questioning. There were no cranial nerve, motor, or sensory deficits, or abnormal reflexes or movements. Examination of the head, skin, chest, cardiovascular system, abdomen, and extremities was normal. Serum sodium was 136 mmol/L, creatinine 1.2 mg/dL, calcium 9.3 mg/dL, and glucose 81 mg/dL; other routine blood chemistries were normal. Her white blood cell (WBC) count was 7100/L, hemoglobin 9.2 g/dL with normal MCV, and platelet count 275,000/L. INR was 3.4, and liver function tests were normal. CT of the brain demonstrated no evidence of acute pathology.
Given that her laboratory results (aside from the hemoglobin) and CT were essentially normal, the most common etiology of a recurrent encephalopathy would be a toxic exposure including drugs, alcohol, and environmental toxins or poisons. A comprehensive serum drug screen, including heavy metals, could follow a basic urinary screen for drugs of abuse; specific etiologies may be suggested by patterns of injury seen on MRI such as those seen with carbon monoxide or methanol exposure. Other recurrent metabolic processes include the porphyrias and relapsing inflammatory disorders, which could be entertained if further diagnostics are unrevealing.
An EEG is warranted at this point and is a test that is underutilized in the workup of altered mental status. Patients who have a spell and do not quickly awaken should be considered to be in nonconvulsive status epilepticus until proven otherwise. This can be easily identified on the EEG and is an important entity to recognize quickly. Additional findings on EEG may suggest focal cerebral dysfunction (such as that following a seizure or acute unilateral injury), diffuse encephalopathy (eg, triphasic waves), or fairly specific diagnoses (eg, periodic lateralized epileptiform discharges from the temporal lobes in suspected herpes simplex meningoencephalitis). While the CT of the brain is a reasonable initial screen, MRI is more sensitive for structural disease and should be obtained if no etiology is rapidly identified.
Finally, acute infectious etiologies such as abscess, encephalitis, or meningoencephalitis need to be excluded via lumbar puncture. Spinal fluid examination also can be helpful in the consideration of inflammatory and autoimmune disorders.
Over the next several hours, while still in the emergency department, she became increasingly obtunded, to the point that she was unresponsive to all stimuli. No seizure activity was witnessed, her vital signs were unchanged, and no medications had been administered. She was urgently transferred to a tertiary care center, where, at the time of arrival, she was obtunded and nonverbal, and opened her eyes only to noxious stimuli. She would withdraw all 4 extremities in response to pain. Pupils were 2 mm and symmetrically reactive. Corneal reflexes were normal, and her gag reflex was diminished. Motor tone was decreased in all 4 extremities. No fasciculations were noted. Deep tendon reflexes were present but symmetrically diminished throughout, and Babinski testing demonstrated a withdrawal response bilaterally.
Coma is a state of profound unconsciousness where the patient is unarousable and unaware of her surroundings. Coma can result either from bihemispheric dysfunction or diffuse injury to the reticular activating system in the brainstem, and the physical examination should focus on distinguishing between these 2 sites. Because the nuclei of cranial nerves III through XII (excepting XI) reside in the brainstem, the coma examination emphasizes testing the cranial nerves; although all cranial nerves are not tested in this patient, the ones that are appear to be normal, making bihemispheric dysfunction most likely. Bihemispheric coma most commonly results from diffuse toxic or metabolic etiologies such as intoxication or hepatic encephalopathy, but it can also be caused by bilateral structural lesions (including the bilateral thalami) or ongoing seizure activity.
Although an EEG remains the key test in this patient given her past history and an MRI would prove extremely useful, her deterioration warrants a workup for CNS infection. Since the head CT was negative, it would be prudent to proceed with urgent lumbar puncture (although it should never be performed in a patient with significant coagulopathy due to risks of hemorrhage leading to spinal cord injury). She should be covered empirically with broad spectrum meningeal‐dose antibiotics, including acyclovir, until the results of the spinal fluid examination are known, given that bacterial meningitis and herpes meningoencephalitis carry a high morbidity and mortality if not treated promptly.
Routine blood tests were similar to her labs at the referring emergency room. Ammonia level was 10 mol/L. Urine toxicology screen was negative, and blood tests for ethanol, salicylates, lithium, and acetaminophen were negative. Chest X‐ray and urinalysis were normal, and electrocardiogram was notable only for a sinus tachycardia. Cultures of the blood were obtained and the patient was admitted to the intensive care unit.
Levetiracetam, vancomycin, piperacillin‐tazobactam, and acyclovir were initiated. A lumbar puncture was performed without reversing the anticoagulation, and the procedure was traumatic. The cerebrospinal fluid was bloody, with a clear supernatant. Cell count demonstrated a red blood cell (RBC) count of 1250/L and a WBC count of 9/L, with a WBC differential of 42% neutrophils, 48% lymphocytes, and 8% monocytes. The cerebrospinal fluid (CSF) glucose was 62 mg/dL (with a serum glucose of 74 mg/dL) and protein 41 mg/dL. The CSF Gram stain demonstrated no organisms, and fluid was sent for routine culture and polymerase chain reaction (PCR) to detect herpes simplex virus (HSV). A neurology consultation was urgently requested.
As mentioned, it would have been more appropriate to reverse the patient's anticoagulation prior to lumbar puncture. The absence of xanthochromia suggests that the RBCs seen in the sample were introduced at the time of the lumbar puncture, arguing against a hemorrhagic disorder of the CNS (occasionally seen with herpes simplex encephalitis) or spinal fluid (eg, subarachnoid hemorrhage).
A reasonable rule of thumb to correct for the number of RBCs in a traumatic lumbar puncture is to allow 1 WBC for every 700 RBCs/L. Given this conversion, there are still too many WBCs in this sample, indicating a mild pleocytosis that is approximately one‐half neutrophilic and one‐half lymphocytic. This profile is nonspecific and can occur with a variety of conditions including stroke, seizure, inflammatory disorders, and infections, including viruses such as West Nile virus.
While coverage with acyclovir and broad‐spectrum antibacterials is appropriate, it should be noted that piperacillin‐tazobactam has poor CSF penetration and therefore is not a good choice for empiric coverage of CNS infections.
The neurologist's examination additionally noted multifocal myoclonus with noxious stimuli, most prominent in the face and toes. An urgent EEG demonstrated continuous, slow, generalized triphasic wave activity (Figures 1 and 2); no epileptiform discharges were seen. The erythrocyte sedimentation rate (ESR) was 66 mm/hour (normal, 0‐30), and tests for antinuclear antibodies, serum levetiracetam level, and thyroid function studies were ordered.
Stimulus‐evoked multifocal myoclonus is a general marker of encephalopathy found in many conditions, including hepatic and renal failure, drug intoxication (eg, opiates), neurodegenerative disorders (eg, Creutzfeldt‐Jakob disease [CJD]), and postanoxic injury, the latter of which is termed the Lance‐Adams syndrome.
Triphasic waves on EEG, while commonly associated with hepatic encephalopathy, have a similarly broad differential diagnosis, although in a comatose patient, they must first and foremost be distinguished from the repetitive discharges characteristic of nonconvulsive status epilepticus. In addition to hepatic and renal failure, triphasic waves have also been described in medication toxicity (especially with anticonvulsants, lithium, and cephalosporins), CNS infections (including Lyme disease and West Nile virus), strokes involving the bilateral thalami (usually from deep venous thrombosis), inflammatory disorders (such as Hashimoto's encephalopathy [HE]), and neurodegenerative diseases. It is important to remember that a single EEG does not exclude the possibility of an episodic ictal disorder and longer‐term monitoring would be required to definitively exclude seizures.
At this point, although the myoclonus and triphasic waves most commonly would indicate a toxic/metabolic process, the elevated ESR and CSF pleocytosis argue for an inflammatory or infectious condition. An MRI remains the next most useful test to guide further workup because many such conditions have distinct signatures on MRI.
The following day, she was noted to have periods of alertnessopening her eyes and following some commandsbut at other times she was difficult to arouse or obtunded. Tremulous movements and sporadic myoclonic jerks continued but no focal neurologic signs were found. Although there was increased muscle tone throughout, she was intermittently seen moving her limbs spontaneously, but not to command. No new findings were appreciated on routine laboratory tests. Antinuclear antibody testing was negative. Serum levetiracetam level was 23.5 g/mL (reference range, 545). Serum thyroid‐stimulating hormone was less than 0.005 U/mL, but free T3 was 3.5 pg/mL (normal, 1.8‐4.6) and free T4 was 2.0 ng/dL (normal, 0.71.8). An MRI of the brain was compromised by motion artifact but no significant abnormalities were appreciated.
At this point, a family member in another state disclosed that the patient had also been hospitalized 2 months previously while visiting him. Her chief complaint had been shortness of breath. The records were obtained; a cardiac catheterization had revealed nonobstructive coronary disease, and medical management was recommended. The notes mentioned that during the hospitalization she developed altered mental status with disorientation and shaking. CT and MRI of the brain had been unremarkable. The confusion was not explained, but she was discharged in good condition, alert and fully‐oriented.
The additional history confirms a relapsing encephalopathy, now with at least 3 occurrences. The most common etiologies in the face of a normal MRI and basic labs would be recurrent intoxication or exposures, but the inflammatory CSF profile and elevated ESR are not consistent with this. A variety of inflammatory disorders can present with recurrent encephalopathy, including demyelinating diseases and neurosarcoidosis. Some systemic rheumatologic conditions, such as systemic lupus erythematosus, can present with relapsing neurologic symptoms due to seizures, vasculitis, or cerebritis. Vasculitis would fit this picture as well, except for the normal findings on 2 MRIs. In a patient with such dramatic symptoms of neurologic dysfunction, one would expect to see changes on the MRI of cerebral inflammation with probable ischemia.
Therefore, given the CSF, ESR, clinical course, and unrevealing MRI and EEG, the most likely group of disorders responsible would be the nonvasculitic autoimmune meningoencephalitides, which present with recurrent encephalopathy and feature spontaneous remissions and/or often‐dramatic responses to corticosteroids. Key disorders in this category include Sjogren's disease, lupus, and steroid responsive encephalopathy associated with autoimmune thyroiditis (Hashimoto's encephalopathy). The latter condition is the most common of the group and is suggested by the abnormal thyroid‐stimulating hormone testing, although it may occur in the setting of normal thyroid function. The diagnosis can be confirmed with thyroperoxidase and thyroglobulin antibody testing.
Three days into the hospitalization, her mental status had gradually improved such that she was more consistently awake and oriented to person and place, and she was transferred to a regular nursing unit. Final results from the CSF and blood cultures were negative, as was PCR for HSV. The antimicrobials were discontinued. Routine serum chemistries continued to be unremarkable. Additional studies recommended by the neurologist demonstrated an antithyroperoxidase antibody concentration of 587.1 IU/mL (normal, <5), and antithyroglobulin antibody level of 52.2 IU/mL (normal, <10).
These results confirm the diagnosis of HE which, in addition to its presentation as a recurrent illness, is an important treatable cause of dementia and should be considered in young patients, those with autoimmune and thyroid disorders, and those whose dementia is rapidly progressive. Most cases are thought to be steroid‐responsive, but some studies have defined the disorder based on this responsiveness, resulting in some nonresponders likely being overlooked.
A trial of corticosteroids should be considered if the patient does not quickly return to baseline given the potential morbidities associated with prolonged altered mental status to this degree. Whether initiation of chronic immunosuppression could prevent these attacks in the future is unclear from the literature but should be considered given the recurrent, dramatic presentation in this patient.
A diagnosis of HE was made, and she was prescribed corticosteroids. Twenty‐four hours later, she was alert and fully‐oriented. She was discharged to home on prednisone and seen in follow‐up in neurology clinic 1 month later. She had had no further episodes of confusion or stupor, but because of steroid‐induced hyperglycemia, her corticosteroids were decreased and mycophenolate mofetil added for chronic immunosuppression. Four months after discharge she was neurologically stable but continued to struggle with the adverse effects of chronic corticosteroid treatment.
COMMENTARY
HE is an uncommon condition that can present with a rapidly progressive decline and should be considered in patients who present with recurrent mental status change in the setting of normal imaging studies and routine laboratory results. The entity was initially described by Lord William Russell Brain in 1966, and in the most recent terminology is known as steroid‐responsive encephalopathy associated with autoimmune thyroiditis (SREAT).1 It is characterized by an acute or subacute encephalopathy associated with thyroid autoimmunity. Patients typically present with fluctuating symptoms, episodes of confusion, alterations of consciousness, and rapid cognitive decline.2 Common features include myoclonus, tremor, ataxia, speech disturbance, stroke‐like episodes, increased muscle tone, neuropsychiatric manifestations, and seizures, that in some cases may progress to status epilepticus.3, 4
Although serum antithyroglobulin and antithyroperoxidase antibodies are elevated in HE, their presence is thought to be an epiphenomenon of the condition rather than the direct cause. Supporting this are the facts that the incidence of encephalopathy is not increased in patients with established autoimmune thyroiditis, and the presence of antithyroid antibodies ranges from 5% to 20% in the general population.2, 5 There is also no evidence that thyroid antibodies directly react with brain tissue, and the levels of these antibodies do not correlate with either neurologic manifestations or clinical improvement.2, 4, 5 As HE has been reported in patients with euthyroidism, hypothyroidism, and hyperthyroidism (with hypothyroidismeither subclinical or activemost common), it is also unlikely that the level of thyroid hormones play a role in the etiology of this disease.2, 4, 6
The etiology and pathogenesis of HE are unclear, although an immune‐mediated process is generally implicated, either from an inflammatory vasculitis or as a form of acute disseminated encephalomyelitis.7‐9 Global hypoperfusion on single‐photon emission computed tomography (SPECT) studies has also been reported.10, 11 Patients with HE may have nonspecific evidence of inflammation, including an elevated ESR, CRP, and CSF protein.12 Other laboratory abnormalities may include a mild elevation of liver aminotransferase levels; renal impairment has also been reported in a few cases of HE in the form of glomerulonephritis, and may be related to deposition of immune complexes containing thyroglobulin antigen.6, 12‐14 MRI of the brain is normal or nonspecific in most cases, and the EEG most commonly shows diffuse slowing.
The differential for a rapidly progressive cognitive decline includes CJD, CNS vasculitis, paraneoplastic syndromes, and autoimmune and subacute infectious encephalopathies. In patients with CJD, T2‐weighted imaging may show hyperintense signals in the basal ganglia, while diffusion‐weighted sequences may reveal changes in the cortical ribbon and bilateral thalami.15 In CNS vasculitis, the imaging findings are variable and range from discrete areas of vascular infarcts to hemorrhagic lesions.16 In paraneoplastic and autoimmune encephalopathies (excluding HE), MRI often shows nonenhancing signal intensity changes in the mesial temporal lobes.12 This patient had repeatedly normal MRI studies of the brain, which in combination with the history of tremor, myoclonus, seizures, and interval return to baseline status, helped point to the diagnosis of HE.
Different approaches to treatment of HE have been recommended. As the acronym SREAT suggests, patients typically respond dramatically to high‐dose steroid therapy. Although a number of patients also improve spontaneously, up to 60% of patients experience a relapsing course and require chronic immunosuppressive agents for maintenance therapy, including long‐term steroids and azathioprine.2, 17 Treatment with plasma exchange and intravenous immune globulin have also been reported, but with mixed results.18, 19 Due to her history of multiple relapses, the patient was placed on mycophenolate mofetil for additional maintenance immunosuppression, as her corticosteroid dose was reduced due to adverse effects.
Acute mental status change is a potentially emergent situation that must be evaluated with careful history and studies to exclude life‐threatening metabolic, infectious, and vascular conditions. This patient presented similarly on 2 prior occasions, and each time her physician team evaluated what appeared to be a new onset of altered consciousness, reaching a plausible but ultimately incorrect diagnosis. The patient's third presentation was finally the charm, as her physicians learned of the repeated history of a confusional state, and in particular the return to baseline status, allowing them to create a differential that focused on etiologies of relapsing encephalopathy and make the correct diagnosis.
Key Points
-
Recurrent acute or subacute cognitive deterioration invokes a differential diagnosis of toxic/metabolic disorders and unusual inflammatory conditions.
-
The nonvasculitic autoimmune encephalopathies are a group of uncommon conditions characterized by nonspecific findings of inflammation and generally unremarkable CNS imaging studies.
-
HE, or SREAT, is the most common of these conditions, and is notable for mental status changes, various findings of increased muscular tone, thyroid autoimmunity, and generally a dramatic response to corticosteroids.
- , , .Hashimoto's disease and encephalopathy.Lancet.1966;2:512–514.
- , , .Hashimoto encephalopathy: syndrome or myth?Arch Neurol.2003;60:164–171.
- , , .Pisani F. Recurrent status epilepticus as the main feature of Hashimoto's encephalopathy.Epilepsy Behav.2006;8:328–330.
- , , , et al.Steroid‐responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol.2006;63:197–202.
- , , , , .Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment.J Neurol.1996;243:585–593.
- , , , , .Hashimoto's encephalopathy: a steroid‐responsive disorder associated with high anti‐thyroid antibody titers‐report of 5 cases.Neurology.1991;41:228–233.
- , , , , .Hashimoto encephalopathy: a brainstem vasculitis?Neurology.2000;54:769–770.
- , , , , .Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): A reversible form of encephalopathy.Neurology.1999;53:1579–1581.
- , , , .Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus.Neurology.2003;61:1124–1126.
- , , .Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism.Neurology.1997;49:623–626.
- , , , , .Hashimoto's encephalopathy: clinical, SPECT and neurophysiologic data.QJM.2003;96:455–457.
- , , .Autoimmune Encephalopathies.The Neurologist.2007;13:140–147.
- , , , , , .Thyroid antigen‐antibody nephritis.Clin Immunol Immunopathol1976;6:341–346.
- , , .Immune complex glomerulonephritis mediated by thyroid antigens.Arch Pathol Lab Med1978;102:530–533.
- , , , et al.Diffusion‐weighted MR imaging of early‐stage Creutzfeldt‐Jakob disease: typical and atypical manifestations.Radiographics.2006;26:S191–S204.
- , , , , .CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.AJNR Am J Neuroradiol.1999;20:75–85.
- , .Long‐Term Treatment of Hashimoto's Encephalopathy.J Neuropsychiatry Clin Neurosci.2006;18:14–20.
- , .Hashimoto's encephalopathy: steroid resistance and response to intravenouc immunoglobulins.J Neurol Neurosurg Psychiatry.2005;76:455–456.
- , .Hashimoto's encephalopathy responding to plasmapheresis.J Neurol Neurosurg Psychiatry.2001;70:132.
- , , .Hashimoto's disease and encephalopathy.Lancet.1966;2:512–514.
- , , .Hashimoto encephalopathy: syndrome or myth?Arch Neurol.2003;60:164–171.
- , , .Pisani F. Recurrent status epilepticus as the main feature of Hashimoto's encephalopathy.Epilepsy Behav.2006;8:328–330.
- , , , et al.Steroid‐responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol.2006;63:197–202.
- , , , , .Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment.J Neurol.1996;243:585–593.
- , , , , .Hashimoto's encephalopathy: a steroid‐responsive disorder associated with high anti‐thyroid antibody titers‐report of 5 cases.Neurology.1991;41:228–233.
- , , , , .Hashimoto encephalopathy: a brainstem vasculitis?Neurology.2000;54:769–770.
- , , , , .Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): A reversible form of encephalopathy.Neurology.1999;53:1579–1581.
- , , , .Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus.Neurology.2003;61:1124–1126.
- , , .Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism.Neurology.1997;49:623–626.
- , , , , .Hashimoto's encephalopathy: clinical, SPECT and neurophysiologic data.QJM.2003;96:455–457.
- , , .Autoimmune Encephalopathies.The Neurologist.2007;13:140–147.
- , , , , , .Thyroid antigen‐antibody nephritis.Clin Immunol Immunopathol1976;6:341–346.
- , , .Immune complex glomerulonephritis mediated by thyroid antigens.Arch Pathol Lab Med1978;102:530–533.
- , , , et al.Diffusion‐weighted MR imaging of early‐stage Creutzfeldt‐Jakob disease: typical and atypical manifestations.Radiographics.2006;26:S191–S204.
- , , , , .CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.AJNR Am J Neuroradiol.1999;20:75–85.
- , .Long‐Term Treatment of Hashimoto's Encephalopathy.J Neuropsychiatry Clin Neurosci.2006;18:14–20.
- , .Hashimoto's encephalopathy: steroid resistance and response to intravenouc immunoglobulins.J Neurol Neurosurg Psychiatry.2005;76:455–456.
- , .Hashimoto's encephalopathy responding to plasmapheresis.J Neurol Neurosurg Psychiatry.2001;70:132.