User login
Autoimmune pancreatitis: A mimic of pancreatic cancer
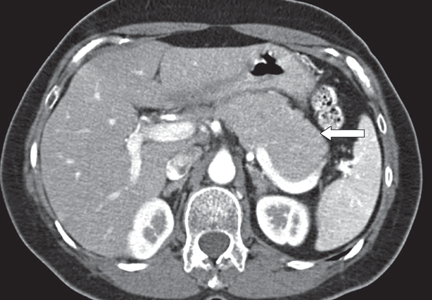
A 66-year-old Korean man presented with a 2-week history of progressive jaundice, mild epigastric discomfort, and a weight loss of 12 lb. His serum bilirubin level was 5.8 mg/dL (reference range 0.0–1.5), and his alkaline phosphatase level was 325 U/L (20–120). Computed tomography (CT) revealed a 3-cm mass in the head of the pancreas.
Exploratory laparotomy revealed a fibrotic pancreas with a palpable mass in the pancreatic head. The mass was unresectable, as it was adhering to the portal vein. A choledochoduodenostomy (anastamosis of the common bile duct to the duodenum) was created for palliation of jaundice. Intraoperative core biopsy revealed destruction of the pancreatic acinar architecture by marked lymphoplasmacytic inflammation and lymphocytic and obliterative venulitis, consistent with autoimmune pancreatitis.
Immediately after surgery, his serum immunoglobulin G4 (IgG4) level was 380 mg/dL (reference range 1–112). His bilirubin and alkaline phosphatase values came down into the normal range in the immediate postoperative period, and his jaundice resolved after a few days.
A CHRONIC INFLAMMATORY CONDITION
Autoimmune pancreatitis is a chronic inflammatory condition with distinct clinical, radiographic, and histologic features.
Sarles et al,1 in 1961, were first to propose that autoimmunity may be a factor in chronic pancreatitis. Three decades later, autoimmune pancreatitis was codified as a separate disease on the basis of a case report of a patient with serum elevations of IgG and gamma globulin, pancreatic duct narrowing, lymphocytic infiltration, fibrosis, and a marked response to steroid therapy.2 Yet its pathogenesis remains poorly understood.
Extrapancreatic manifestations include sclerosing sialadenitis, sclerosing cholangitis, and retroperitoneal fibrosis.
Of note, autoimmune pancreatitis can mimic pancreatic adenocarcinoma clinically and radiographically. One must differentiate between the two disorders to prevent unnecessary surgery or delay in corticosteroid therapy.
RATES ARE POORLY DEFINED
The exact prevalence and incidence of autoimmune pancreatitis remain poorly defined. Most of the initial epidemiologic data have come from Japan and Korea. The prevalence was 0.7 per 100,000 patients in a survey of the Japanese population.3 Further studies are needed to ascertain its incidence and prevalence in the United States.
In patients with chronic pancreatitis, the estimated prevalence is between 4.6% and 6%, and 11% in patients undergoing pancreatic resection for suspected pancreatic cancer.4,5
Autoimmune pancreatitis appears to be a disease of the elderly, as most patients are more than 50 years old at diagnosis. Twice as many men as women are affected.5 Many patients have no history of alcohol abuse or other traditional risk factors for chronic pancreatitis.
CLINICAL PRESENTATION: PAINLESS JAUNDICE, WEIGHT LOSS

The most common clinical presentation is obstructive jaundice with little or no abdominal pain. In one series,4 65% of patients presented with painless jaundice secondary to biliary obstruction. Obstructive acute pancreatitis can occur, due to inflammatory strictures of the main pancreatic duct.
Weight loss results from impaired digestion and decreased appetite. Autoimmune pancreatitis is complicated by pancreatic exocrine insufficiency in 88% of cases6 and by endocrine dysfunction in 67%.3
Many patients have extrapancreatic lesions such as sclerosing sialadenitis, retroperitoneal fibrosis, and autoimmune sclerosing cholangitis.7 The cholangiographic appearance of autoimmune sclerosing cholangitis may resemble that of primary sclerosing cholangitis or cholangiocarcinoma. Less common extrapancreatic findings include interstitial nephritis and mediastinal adenopathy. These extrapancreatic findings do not always coincide with pancreatic inflammation. The histopathologic findings in extrapancreatic lesions parallel those in the pancreas.8
The patient described at the beginning of this article had several of these features, including painless jaundice, weight loss, elevated alkaline phosphatase, and an inflammatory pancreatic mass.
DIAGNOSIS IS IMPROVING
Laboratory findings
Serum amylase and lipase are neither sensitive nor specific for autoimmune pancreatitis. Usually, their values are within normal limits or only mildly elevated.
A cholestatic pattern of elevation (elevated alkaline phosphatase and bilirubin, with normal or only slightly elevated alanine and aspartate aminotransferases) is found in patients with an inflammatory mass in the pancreatic head and in those with autoimmune sclerosing cholangitis. In one series,4 pancreatic enzymes were elevated in only 3 (13%) of 17 cases, while cholestasis was present in 16 (94%).
Gamma globulin, total IgG, and IgG4 are commonly elevated in autoimmune pancreatitis. Serum IgG4 is considered the most sensitive and specific marker and is elevated in 63% to 94% of patients with autoimmune pancreatitis.4,15–17 Several studies found the diagnostic accuracy, sensitivity, and specificity to be highest (> 90%) when a cut point of 135 mg/dL was used.17,18 A subsequent study 15 revealed a sensitivity of 76% and a specificity of 93% using the same cut point. Recall that the IgG4 level in our patient was 380 mg/dL.
Autoantibodies that are elevated in autoimmune pancreatitis include antilactoferrin antibodies and anticarbonic anhydrase II antibodies. 19 Both are “organ-specific”: the former are found in pancreatic acinar cells, and the latter are found in ductal cells. The sensitivity of both antibodies is greater than 50% in patients with autoimmune pancreatitis. However, they are not often measured, since testing for them is not widely available.20,21
Antinuclear antibody and rheumatoid factor are also associated with autoimmune pancreatitis but are not very specific.
Radiographic findings
The most common radiographic feature is diffuse enlargement of the entire pancreas. The appearance of the gland is often described as “sausage-like,” a feature best seen with CT and magnetic resonance imaging (MRI).
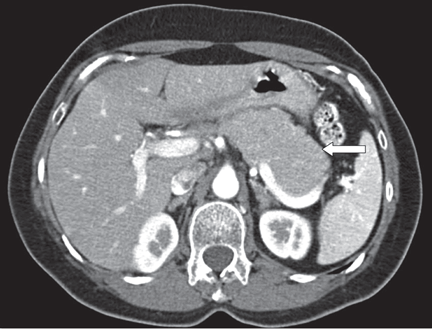
A well-defined capsule-like rim surrounding the pancreas is another common feature.23 This rim-enhancement is hypointense on T2 MRI, suggesting the presence of peripheral inflammation and fibrosis.
Calcifications and pseudocysts are rarely seen in autoimmune pancreatitis.
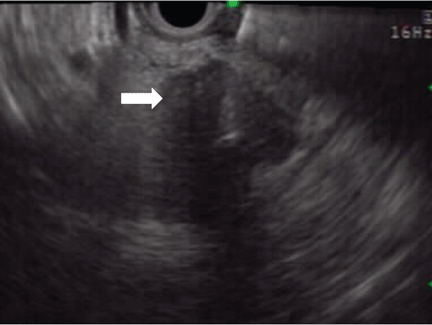
On ultrasonography, the involved pancreatic parenchyma appears hypoechoic, consistent with edema.
Endoscopic retrograde cholangiopancreatography
ERCP or magnetic resonance cholangiopancreatography may reveal segmental or diffuse narrowing of the main pancreatic duct.24,25 Bile-duct strictures may occur throughout the biliary tree.23
Autoimmune pancreatitis with biliary involvement must be distinguished from primary sclerosing cholangitis because the former responds to corticosteroid treatment. Cholangiographic features in primary sclerosing cholangitis include band-like strictures and a beaded or “pruned-tree” appearance, while autoimmune pancreatitis more commonly produces long strictures with prestenotic dilatation.26
ERCP allows temporary stents to be placed in obstructed segments of the biliary tree to open them up in the setting of acute cholangitis.
Biopsy guided by endoscopic ultrasonography
Some have proposed using endoscopic ultrasonography to guide biopsy in cases of suspected autoimmune pancreatitis.27,28
Fine-needle aspiration biopsy, guided by endoscopic ultrasonography, is frequently used to rule out adenocarcinoma. However, its yield for cancer is not perfect (about 70%–90%), so a negative biopsy does not rule out cancer. Further, autoimmune pancreatitis is rare, so a patient with a negative finding on fine-needle aspiration biopsy is still more likely to have cancer than autoimmune pancreatitis. In this case, the negative study should be combined with other information (eg, IgG4) to decide whether empiric treatment should be given.
Core biopsy, also guided by endoscopic ultrasonography, collects a greater amount of tissue for analysis and may allow the histologic diagnosis of autoimmune pancreatitis, but it carries a greater risk of bleeding. Also, its yield may be lower than initially thought. In one series, only 26% of ultrasonographically guided core samples from patients with confirmed autoimmune pancreatitis had diagnostic histologic features.29
New immunohistologic techniques are being developed to increase the yield from cytologic and tissue specimens.
Histopathologic findings

Histologic evaluation remains the gold standard for diagnosis. The histologic diagnosis can be made in patients who have any or all of the following three most common histologic features of autoimmune pancreatitis10,23,31–33:
- Parenchymal and often periductal lymphoplasmacytic infiltration, which is typically florid in intensity
- Storiform fibrosis
- Obliterative phlebitis.
The histologic findings in our patient included lymphoplasmacytic infiltration and obliterative phlebitis, which were essential to establishing the diagnosis. In a series of 53 patients, parenchymal inflammation with periductal lymphoplasmacytic accentuation was found in all of them.33
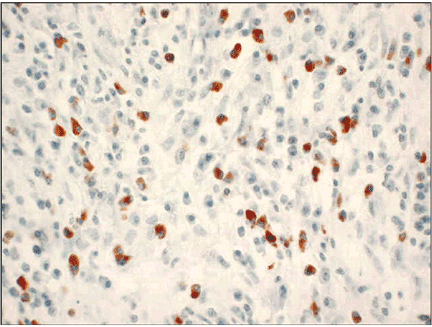
Biopsy of extrapancreatic sites, including the bile ducts and major duodenal papilla, may also facilitate the diagnosis.34,35 In a recent study,34 80% of autoimmune pancreatitis patients with pancreatic head involvement had significant numbers of IgG4-positive cells on biopsy of the major duodenal papilla. Biopsy of the periampullary duodenum may be a safer alternative to guided fine-needle aspiration or core biopsy.
In addition to lymphocytes, the inflammatory infiltrates in autoimmune pancreatitis may contain macrophages, mast cells, neutrophils, and eosinophils. Nonnecrotizing granulomas are occasionally seen, including periductal granulomas.
Fibrosis. Ductal luminal destruction can be seen in conjunction with fibrosis that thickens the duct wall and forms interlobular septa.33 Fibrosis may also affect the acinar tissue and produce profound lobular atrophy. In severe cases, the fibrotic changes can encompass large areas, with myofibroblasts arranged in a storiform pattern resembling an inflammatory pseudotumor.36
Phlebitis. The vascular changes in autoimmune pancreatitis have been underemphasized relative to the pancreatic parenchymal fibroinflammatory changes. Venulitis is seen mainly in small and medium-size pancreatic and peripancreatic veins. The inflammatory response and fibrosis disrupt the venous endothelium and often result in obliterative phlebitis.
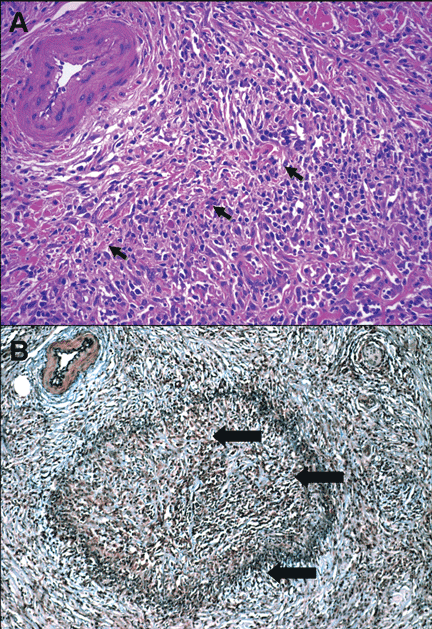
However, Movat staining may not be available if an operative frozen section is being analyzed. In these cases, the venous lesions can be found by localizing the paired arteries, which are usually entirely normal and readily evident. If paired veins are not seen in this manner, a high level of suspicion should be raised for autoimmune pancreatitis with lymphocytic vein destruction.
CORTICOSTEROIDS ARE EFFECTIVE
Our patient’s jaundice temporarily resolved after his biliary bypass operation. If the diagnosis had been made earlier, he could have been treated with corticosteroids.
Corticosteroids have been used to treat autoimmune pancreatitis, with great success. (However, autoimmune pancreatitis occasional resolves spontaneously and stays in remission without corticosteroids.) A common regimen is oral prednisone 40 mg/day for 4 weeks and then tapered by 5 mg every 1 to 2 weeks. Patients who have a delayed response may receive long-term maintenance corticosteroid therapy (2.5–5 mg of oral prednisone).38–40
The radiographic and laboratory abnormalities typically resolve promptly with steroid therapy. A radiographic response is seen as early as 2 to 3 weeks, with normalization occurring in 4 to 6 weeks.40 Serum IgG4 levels decrease concurrently.38
Between 36% and 60% of patients with diabetes and autoimmune pancreatitis have better insulin secretion and glycemic control once corticosteroid therapy is started.3,6,38,40 Fifty percent of patients with exocrine insufficiency have functional improvement after corticosteroid therapy.6
Extrapancreatic lesions also improve with therapy.40,41 Obstructive jaundice may require endoscopic placement of a temporary biliary stent, but after a few weeks of steroid therapy the stent can usually be removed.
The decision to treat with corticosteroids is usually based on symptoms, imaging features (stricture or mass), a low suspicion of cancer (eg, negative biopsy), and an elevated IgG4. A histologic diagnosis of autoimmune pancreatitis is usually not available or required but may be sought through endoscopic ultrasonography-guided core biopsy or laparoscopic biopsy if the diagnosis is in doubt.
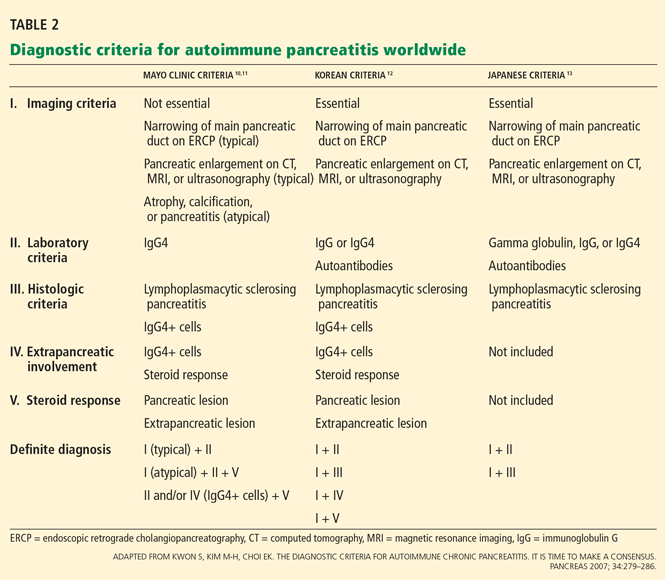
Another reasonable approach is an empiric trial of corticosteroids, reassessing the symptoms and repeating the imaging tests after 1 to 2 months. In fact, a response to corticosteroids is a component of most diagnostic criteria (Table 2).
Recurrence rates range from 6% to 32%.4,33,39,42,43 Patients who relapse after initial corticosteroid therapy may be treated again with prednisone in high doses (40 mg/day).38,41 Immunomodulatory therapy has been used successfully to treat relapsed disease in a single reported series: seven patients received either azathioprine (Imuran) 2 mg/kg daily or mycophenolate mofetil (Cell-Cept) 750 mg twice daily, and all remained in complete remission at a median follow-up of 6 months with no adverse events.44
In cases that fail to respond to corticosteroids, the diagnosis of autoimmune pancreatitis should be re-evaluated and surgery should be considered to look for cancer.
CASE CONTINUED
Our patient felt well at his 2-month follow-up visit. However, his serum alkaline phosphatase had increased to 649 U/L, and his serum IgG4 had increased to 980 mg/dL.
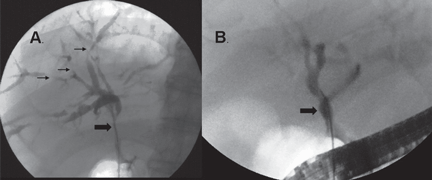
ERCP repeated 6 weeks later showed that the hilar stricture had completely resolved, and the intrahepatic strictures had markedly improved (Figure 6). His serum alkaline phosphatase level was now 73 U/L, and his serum IgG4 was 231 mg/dL.
Almost 2 years after starting corticosteroid therapy, the patient has remained in good control and the prednisone has been tapered off completely. His latest laboratory values are alkaline phosphatase 70 U/L and IgG4 46 mg/dL.
- Sarles H, Sarles JC, Muratore R, Guien C. Chronic inflammatory sclerosis of the pancreas—an autonomous pancreatic disease? Am J Dig Dis 1961; 6:688–698.
- Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci 1995; 40:1561–1568.
- Nishimori I, Tamakoshi A, Kawa S, et al; Research Committee on Intractable Pancreatic Diseases, the Ministry of Health and Welfare of Japan. Influence of steroid therapy on the course of diabetes mellitus in patients with autoimmune pancreatitis: findings from a nationwide survey in Japan. Pancreas 2006; 32:244–248.
- Kim KP, Kim M, Lee YJ, et al. Clinical characteristics of 17 cases of autoimmune chronic pancreatitis. Korean J Gastroenterol 2004; 43:112–119.
- Finkelberg DL, Sahani D, Deshpande V, Brugge WR. Autoimmune pancreatitis. N Engl J Med 2006; 355:2670–2676.
- Kamisawa T, Egawa N, Inokuma S, et al. Pancreatic endocrine and exocrine function and salivary gland function in autoimmune pancreatitis before and after steroid therapy. Pancreas 2003; 27:235–238.
- Kamisawa T, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Extrapancreatic lesions in autoimmune pancreatitis. J Clin Gastroenterol 2005; 39:904–907.
- Kamisawa T, Nakajima H, Egawa N, Funata N, Tsuruta K, Okamoto A. IgG4-related sclerosing disease incorporating sclerosing pancreatitis, cholangitis, sialadenitis and retroperitoneal fibrosis with lymphadenopathy. Pancreatology 2006; 6:132–137.
- Kwon S, Kim MH, Choi EK. The diagnostic criteria for autoimmune chronic pancreatitis: it is time to make a consensus. Pancreas 2007; 34:279–286.
- Chari ST. Diagnosis of autoimmune pancreatitis using its five cardinal features: introducing The Mayo Clinic’s HISORt criteria. J Gastroenterol 2007; 42( suppl 18):39–41.
- Chari ST, Smyrk TC, Levy MJ, et al. Diagnosis of autoimmune pancreatitis: The Mayo Clinic experience. Clin Gastroenterol Hepatol 2006; 4:1010–1016.
- Kim K-P, Kim M-H, Kim JC, Lee SS, Seo DW, Lee SK. Diagnostic criteria for autoimmune chronic pancreatitis revisited. World J Gastroenterol 2006; 12:2487–2496.
- Okazaki K, Kawa S, Kamisawa T. Clinical diagnostic criteria of autoimmune pancreatitis: revised proposal. J Gastroenterol 2006; 41:626–631.
- Nishimori I, Onishi S, Otsuki M. Review of diagnostic criteria for autoimmune pancreatitis; for establishment of international criteria. Clin J Gastroenterol 2008; 1:7–17.
- Ghazale A, Chari ST, Smyrk TC, et al Value of serum IgG4 in the diagnosis of autoimmune pancreatitis and in distinguishing it from pancreatic cancer. Am J Gastroenterol 2007; 102:1646–1653.
- Hirano K, Kawabe T, Yamamoto N, et al. Serum IgG4 concentrations in pancreatic and biliary diseases. Clin Chim Acta 2006; 367:181–184.
- Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001; 344:732–738.
- Kawa S, Hamano H. Clinical features of autoimmune pancreatitis. J Gastroenterol 2007; 42(suppl 18):9–14.
- Okazaki K, Uchida K, Ohana M, et al. Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology 2000; 118:573–581.
- Frulloni L, Bovo P, Brunelli S, et al. Elevated serum levels of antibodies to carbonic anhydrase I and II in patients with chronic pancreatitis. Pancreas 2000; 20:382–388.
- Nishimori I, Miyaji E, Morimoto K, Nagao K, Kamada M, Onishi S. Serum antibodies to carbonic anhydrase IV in patients with autoimmune pancreatitis. Gut 2005; 54:274–281.
- Irie H, Honda H, Baba S, et al. Autoimmune pancreatitis: CT and MR characteristics. AJR Am J Roentgenol 1998; 170:1323–1327.
- Sahani DV, Kalva SP, Farrell J, et al. Autoimmune pancreatitis: imaging features. Radiology 2004; 233:345–352.
- Horiuchi A, Kawa S, Hamano H, Hayama M, Ota H, Kiyosawa K. ERCP features in 27 patients with autoimmune pancreatitis. Gastrointest Endosc 2002; 55:494–499.
- Kamisawa T, Chen PY, Tu Y, et al. MRCP and MRI findings in 9 patients with autoimmune pancreatitis. World J Gastroenterol 2006; 12:2919–2922.
- Nakazawa T, Ohara H, Sano H, et al. Cholangiography can discriminate sclerosing cholangitis with autoimmune pancreatitis from primary sclerosing cholangitis. Gastrointest Endosc 2004; 60:937–944.
- Farrell JJ, Garber J, Sahani D, Brugge WR. EUS findings in patients with autoimmune pancreatitis. Gastrointest Endosc 2004; 60:927–936.
- Levy MJ, Reddy RP, Wiersema MJ, et al. EUS-guided trucut biopsy in establishing autoimmune pancreatitis as the cause of obstructive jaundice. Gastrointest Endosc 2005; 61:467–472.
- Bang SJ, Kim MH, Kim do H, et al. Is pancreatic core biopsy sufficient to diagnose autoimmune chronic pancreatitis? Pancreas 2008; 36:84–89.
- Scully KA, Li SC, Hebert JC, Trainer TD. The characteristic appearance of non-alcoholic duct destructive chronic pancreatitis: a report of 2 cases. Arch Pathol Lab Med 2000; 124:1535–1538.
- Chu KE, Papouchado BG, Lane Z, Bronner MP. The role of Movat pentachrome stain and immunoglobulin G4 immunostaining in the diagnosis of autoimmune pancreatitis. Mod Pathol 2009; 22:351–358.
- Ectors N, Maillet B, Aerts R, et al. Non-alcoholic duct destructive chronic pancreatitis. Gut 1997; 41:263–268.
- Zamboni G, Luttges J, Capelli P, et al. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch 2004; 445:552–563.
- Hamano H, Kawa S, Uehara T, et al. Immunoglobulin G4-related lymphoplasmacytic sclerosing cholangitis that mimics infiltrating hilar cholangiocarcinoma: part of a spectrum of autoimmune pancreatitis? Gastrointest Endosc 2005; 62:152–157.
- Kamisawa T, Tu Y, Egawa N, Tsuruta K, Okamoto A. A new diagnostic endoscopic tool for autoimmune pancreatitis. Gastrointest Endosc 2008; 68:358–361.
- Notohara K, Burgart LJ, Yadav D, Chari S, Smyrk TC. Idiopathic chronic pancreatitis with periductal lymphoplasmacytic infiltration: clinicopathologic features of 35 cases. Am J Surg Pathol 2003; 27:1119–1127.
- Esposito I, Bergmann F, Penzel R, et al. Oligoclonal T-cell populations in an inflammatory pseudotumor of the pancreas possibly related to autoimmune pancreatitis: an immunohistochemical and molecular analysis. Virchows Arch 2004; 444:119–126.
- Kamisawa T, Okamoto A, Wakabayashi T, Watanabe H, Sawabu N. Appropriate steroid therapy for autoimmune pancreatitis based on long-term outcome. Scand J Gastroenterol 2008; 43:609–613.
- Hirano K, Tada M, Isayama H, et al. Long-term prognosis of autoimmune pancreatitis with and without corticosteroid treatment. Gut 2007; 56:1719–1724.
- Kamisawa T, Yoshiike M, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Treating patients with autoimmune pancreatitis: results from a long-term follow-up study. Pancreatology 2005; 5:234–38.
- Kamisawa T, Okamoto A. Prognosis of autoimmune pancreatitis. J Gastroenterol 2007; 42(suppl 18):59–62.
- Takayama M, Hamano H, Ochi Y, et al. Recurrent attacks of autoimmune pancreatitis result in pancreatic stone formation. Am J Gastroenterol 2004; 99:932–937.
- Wakabayashi T, Kawaura Y, Satomura Y, Watanabe H, Motoo Y, Sawabu N. Long-term prognosis of duct-narrowing chronic pancreatitis: strategy for steroid treatment. Pancreas 2005; 30:31–39.
- Ghazale A, Chari ST. Optimising corticosteroid treatment for autoimmune pancreatitis. Gut 2007; 56:1650–1652.
A 66-year-old Korean man presented with a 2-week history of progressive jaundice, mild epigastric discomfort, and a weight loss of 12 lb. His serum bilirubin level was 5.8 mg/dL (reference range 0.0–1.5), and his alkaline phosphatase level was 325 U/L (20–120). Computed tomography (CT) revealed a 3-cm mass in the head of the pancreas.
Exploratory laparotomy revealed a fibrotic pancreas with a palpable mass in the pancreatic head. The mass was unresectable, as it was adhering to the portal vein. A choledochoduodenostomy (anastamosis of the common bile duct to the duodenum) was created for palliation of jaundice. Intraoperative core biopsy revealed destruction of the pancreatic acinar architecture by marked lymphoplasmacytic inflammation and lymphocytic and obliterative venulitis, consistent with autoimmune pancreatitis.
Immediately after surgery, his serum immunoglobulin G4 (IgG4) level was 380 mg/dL (reference range 1–112). His bilirubin and alkaline phosphatase values came down into the normal range in the immediate postoperative period, and his jaundice resolved after a few days.
A CHRONIC INFLAMMATORY CONDITION
Autoimmune pancreatitis is a chronic inflammatory condition with distinct clinical, radiographic, and histologic features.
Sarles et al,1 in 1961, were first to propose that autoimmunity may be a factor in chronic pancreatitis. Three decades later, autoimmune pancreatitis was codified as a separate disease on the basis of a case report of a patient with serum elevations of IgG and gamma globulin, pancreatic duct narrowing, lymphocytic infiltration, fibrosis, and a marked response to steroid therapy.2 Yet its pathogenesis remains poorly understood.
Extrapancreatic manifestations include sclerosing sialadenitis, sclerosing cholangitis, and retroperitoneal fibrosis.
Of note, autoimmune pancreatitis can mimic pancreatic adenocarcinoma clinically and radiographically. One must differentiate between the two disorders to prevent unnecessary surgery or delay in corticosteroid therapy.
RATES ARE POORLY DEFINED
The exact prevalence and incidence of autoimmune pancreatitis remain poorly defined. Most of the initial epidemiologic data have come from Japan and Korea. The prevalence was 0.7 per 100,000 patients in a survey of the Japanese population.3 Further studies are needed to ascertain its incidence and prevalence in the United States.
In patients with chronic pancreatitis, the estimated prevalence is between 4.6% and 6%, and 11% in patients undergoing pancreatic resection for suspected pancreatic cancer.4,5
Autoimmune pancreatitis appears to be a disease of the elderly, as most patients are more than 50 years old at diagnosis. Twice as many men as women are affected.5 Many patients have no history of alcohol abuse or other traditional risk factors for chronic pancreatitis.
CLINICAL PRESENTATION: PAINLESS JAUNDICE, WEIGHT LOSS
The most common clinical presentation is obstructive jaundice with little or no abdominal pain. In one series,4 65% of patients presented with painless jaundice secondary to biliary obstruction. Obstructive acute pancreatitis can occur, due to inflammatory strictures of the main pancreatic duct.
Weight loss results from impaired digestion and decreased appetite. Autoimmune pancreatitis is complicated by pancreatic exocrine insufficiency in 88% of cases6 and by endocrine dysfunction in 67%.3
Many patients have extrapancreatic lesions such as sclerosing sialadenitis, retroperitoneal fibrosis, and autoimmune sclerosing cholangitis.7 The cholangiographic appearance of autoimmune sclerosing cholangitis may resemble that of primary sclerosing cholangitis or cholangiocarcinoma. Less common extrapancreatic findings include interstitial nephritis and mediastinal adenopathy. These extrapancreatic findings do not always coincide with pancreatic inflammation. The histopathologic findings in extrapancreatic lesions parallel those in the pancreas.8
The patient described at the beginning of this article had several of these features, including painless jaundice, weight loss, elevated alkaline phosphatase, and an inflammatory pancreatic mass.
DIAGNOSIS IS IMPROVING
Laboratory findings
Serum amylase and lipase are neither sensitive nor specific for autoimmune pancreatitis. Usually, their values are within normal limits or only mildly elevated.
A cholestatic pattern of elevation (elevated alkaline phosphatase and bilirubin, with normal or only slightly elevated alanine and aspartate aminotransferases) is found in patients with an inflammatory mass in the pancreatic head and in those with autoimmune sclerosing cholangitis. In one series,4 pancreatic enzymes were elevated in only 3 (13%) of 17 cases, while cholestasis was present in 16 (94%).
Gamma globulin, total IgG, and IgG4 are commonly elevated in autoimmune pancreatitis. Serum IgG4 is considered the most sensitive and specific marker and is elevated in 63% to 94% of patients with autoimmune pancreatitis.4,15–17 Several studies found the diagnostic accuracy, sensitivity, and specificity to be highest (> 90%) when a cut point of 135 mg/dL was used.17,18 A subsequent study 15 revealed a sensitivity of 76% and a specificity of 93% using the same cut point. Recall that the IgG4 level in our patient was 380 mg/dL.
Autoantibodies that are elevated in autoimmune pancreatitis include antilactoferrin antibodies and anticarbonic anhydrase II antibodies. 19 Both are “organ-specific”: the former are found in pancreatic acinar cells, and the latter are found in ductal cells. The sensitivity of both antibodies is greater than 50% in patients with autoimmune pancreatitis. However, they are not often measured, since testing for them is not widely available.20,21
Antinuclear antibody and rheumatoid factor are also associated with autoimmune pancreatitis but are not very specific.
Radiographic findings
The most common radiographic feature is diffuse enlargement of the entire pancreas. The appearance of the gland is often described as “sausage-like,” a feature best seen with CT and magnetic resonance imaging (MRI).
A well-defined capsule-like rim surrounding the pancreas is another common feature.23 This rim-enhancement is hypointense on T2 MRI, suggesting the presence of peripheral inflammation and fibrosis.
Calcifications and pseudocysts are rarely seen in autoimmune pancreatitis.
On ultrasonography, the involved pancreatic parenchyma appears hypoechoic, consistent with edema.
Endoscopic retrograde cholangiopancreatography
ERCP or magnetic resonance cholangiopancreatography may reveal segmental or diffuse narrowing of the main pancreatic duct.24,25 Bile-duct strictures may occur throughout the biliary tree.23
Autoimmune pancreatitis with biliary involvement must be distinguished from primary sclerosing cholangitis because the former responds to corticosteroid treatment. Cholangiographic features in primary sclerosing cholangitis include band-like strictures and a beaded or “pruned-tree” appearance, while autoimmune pancreatitis more commonly produces long strictures with prestenotic dilatation.26
ERCP allows temporary stents to be placed in obstructed segments of the biliary tree to open them up in the setting of acute cholangitis.
Biopsy guided by endoscopic ultrasonography
Some have proposed using endoscopic ultrasonography to guide biopsy in cases of suspected autoimmune pancreatitis.27,28
Fine-needle aspiration biopsy, guided by endoscopic ultrasonography, is frequently used to rule out adenocarcinoma. However, its yield for cancer is not perfect (about 70%–90%), so a negative biopsy does not rule out cancer. Further, autoimmune pancreatitis is rare, so a patient with a negative finding on fine-needle aspiration biopsy is still more likely to have cancer than autoimmune pancreatitis. In this case, the negative study should be combined with other information (eg, IgG4) to decide whether empiric treatment should be given.
Core biopsy, also guided by endoscopic ultrasonography, collects a greater amount of tissue for analysis and may allow the histologic diagnosis of autoimmune pancreatitis, but it carries a greater risk of bleeding. Also, its yield may be lower than initially thought. In one series, only 26% of ultrasonographically guided core samples from patients with confirmed autoimmune pancreatitis had diagnostic histologic features.29
New immunohistologic techniques are being developed to increase the yield from cytologic and tissue specimens.
Histopathologic findings
Histologic evaluation remains the gold standard for diagnosis. The histologic diagnosis can be made in patients who have any or all of the following three most common histologic features of autoimmune pancreatitis10,23,31–33:
- Parenchymal and often periductal lymphoplasmacytic infiltration, which is typically florid in intensity
- Storiform fibrosis
- Obliterative phlebitis.
The histologic findings in our patient included lymphoplasmacytic infiltration and obliterative phlebitis, which were essential to establishing the diagnosis. In a series of 53 patients, parenchymal inflammation with periductal lymphoplasmacytic accentuation was found in all of them.33
Biopsy of extrapancreatic sites, including the bile ducts and major duodenal papilla, may also facilitate the diagnosis.34,35 In a recent study,34 80% of autoimmune pancreatitis patients with pancreatic head involvement had significant numbers of IgG4-positive cells on biopsy of the major duodenal papilla. Biopsy of the periampullary duodenum may be a safer alternative to guided fine-needle aspiration or core biopsy.
In addition to lymphocytes, the inflammatory infiltrates in autoimmune pancreatitis may contain macrophages, mast cells, neutrophils, and eosinophils. Nonnecrotizing granulomas are occasionally seen, including periductal granulomas.
Fibrosis. Ductal luminal destruction can be seen in conjunction with fibrosis that thickens the duct wall and forms interlobular septa.33 Fibrosis may also affect the acinar tissue and produce profound lobular atrophy. In severe cases, the fibrotic changes can encompass large areas, with myofibroblasts arranged in a storiform pattern resembling an inflammatory pseudotumor.36
Phlebitis. The vascular changes in autoimmune pancreatitis have been underemphasized relative to the pancreatic parenchymal fibroinflammatory changes. Venulitis is seen mainly in small and medium-size pancreatic and peripancreatic veins. The inflammatory response and fibrosis disrupt the venous endothelium and often result in obliterative phlebitis.
However, Movat staining may not be available if an operative frozen section is being analyzed. In these cases, the venous lesions can be found by localizing the paired arteries, which are usually entirely normal and readily evident. If paired veins are not seen in this manner, a high level of suspicion should be raised for autoimmune pancreatitis with lymphocytic vein destruction.
CORTICOSTEROIDS ARE EFFECTIVE
Our patient’s jaundice temporarily resolved after his biliary bypass operation. If the diagnosis had been made earlier, he could have been treated with corticosteroids.
Corticosteroids have been used to treat autoimmune pancreatitis, with great success. (However, autoimmune pancreatitis occasional resolves spontaneously and stays in remission without corticosteroids.) A common regimen is oral prednisone 40 mg/day for 4 weeks and then tapered by 5 mg every 1 to 2 weeks. Patients who have a delayed response may receive long-term maintenance corticosteroid therapy (2.5–5 mg of oral prednisone).38–40
The radiographic and laboratory abnormalities typically resolve promptly with steroid therapy. A radiographic response is seen as early as 2 to 3 weeks, with normalization occurring in 4 to 6 weeks.40 Serum IgG4 levels decrease concurrently.38
Between 36% and 60% of patients with diabetes and autoimmune pancreatitis have better insulin secretion and glycemic control once corticosteroid therapy is started.3,6,38,40 Fifty percent of patients with exocrine insufficiency have functional improvement after corticosteroid therapy.6
Extrapancreatic lesions also improve with therapy.40,41 Obstructive jaundice may require endoscopic placement of a temporary biliary stent, but after a few weeks of steroid therapy the stent can usually be removed.
The decision to treat with corticosteroids is usually based on symptoms, imaging features (stricture or mass), a low suspicion of cancer (eg, negative biopsy), and an elevated IgG4. A histologic diagnosis of autoimmune pancreatitis is usually not available or required but may be sought through endoscopic ultrasonography-guided core biopsy or laparoscopic biopsy if the diagnosis is in doubt.
Another reasonable approach is an empiric trial of corticosteroids, reassessing the symptoms and repeating the imaging tests after 1 to 2 months. In fact, a response to corticosteroids is a component of most diagnostic criteria (Table 2).
Recurrence rates range from 6% to 32%.4,33,39,42,43 Patients who relapse after initial corticosteroid therapy may be treated again with prednisone in high doses (40 mg/day).38,41 Immunomodulatory therapy has been used successfully to treat relapsed disease in a single reported series: seven patients received either azathioprine (Imuran) 2 mg/kg daily or mycophenolate mofetil (Cell-Cept) 750 mg twice daily, and all remained in complete remission at a median follow-up of 6 months with no adverse events.44
In cases that fail to respond to corticosteroids, the diagnosis of autoimmune pancreatitis should be re-evaluated and surgery should be considered to look for cancer.
CASE CONTINUED
Our patient felt well at his 2-month follow-up visit. However, his serum alkaline phosphatase had increased to 649 U/L, and his serum IgG4 had increased to 980 mg/dL.
ERCP repeated 6 weeks later showed that the hilar stricture had completely resolved, and the intrahepatic strictures had markedly improved (Figure 6). His serum alkaline phosphatase level was now 73 U/L, and his serum IgG4 was 231 mg/dL.
Almost 2 years after starting corticosteroid therapy, the patient has remained in good control and the prednisone has been tapered off completely. His latest laboratory values are alkaline phosphatase 70 U/L and IgG4 46 mg/dL.
A 66-year-old Korean man presented with a 2-week history of progressive jaundice, mild epigastric discomfort, and a weight loss of 12 lb. His serum bilirubin level was 5.8 mg/dL (reference range 0.0–1.5), and his alkaline phosphatase level was 325 U/L (20–120). Computed tomography (CT) revealed a 3-cm mass in the head of the pancreas.
Exploratory laparotomy revealed a fibrotic pancreas with a palpable mass in the pancreatic head. The mass was unresectable, as it was adhering to the portal vein. A choledochoduodenostomy (anastamosis of the common bile duct to the duodenum) was created for palliation of jaundice. Intraoperative core biopsy revealed destruction of the pancreatic acinar architecture by marked lymphoplasmacytic inflammation and lymphocytic and obliterative venulitis, consistent with autoimmune pancreatitis.
Immediately after surgery, his serum immunoglobulin G4 (IgG4) level was 380 mg/dL (reference range 1–112). His bilirubin and alkaline phosphatase values came down into the normal range in the immediate postoperative period, and his jaundice resolved after a few days.
A CHRONIC INFLAMMATORY CONDITION
Autoimmune pancreatitis is a chronic inflammatory condition with distinct clinical, radiographic, and histologic features.
Sarles et al,1 in 1961, were first to propose that autoimmunity may be a factor in chronic pancreatitis. Three decades later, autoimmune pancreatitis was codified as a separate disease on the basis of a case report of a patient with serum elevations of IgG and gamma globulin, pancreatic duct narrowing, lymphocytic infiltration, fibrosis, and a marked response to steroid therapy.2 Yet its pathogenesis remains poorly understood.
Extrapancreatic manifestations include sclerosing sialadenitis, sclerosing cholangitis, and retroperitoneal fibrosis.
Of note, autoimmune pancreatitis can mimic pancreatic adenocarcinoma clinically and radiographically. One must differentiate between the two disorders to prevent unnecessary surgery or delay in corticosteroid therapy.
RATES ARE POORLY DEFINED
The exact prevalence and incidence of autoimmune pancreatitis remain poorly defined. Most of the initial epidemiologic data have come from Japan and Korea. The prevalence was 0.7 per 100,000 patients in a survey of the Japanese population.3 Further studies are needed to ascertain its incidence and prevalence in the United States.
In patients with chronic pancreatitis, the estimated prevalence is between 4.6% and 6%, and 11% in patients undergoing pancreatic resection for suspected pancreatic cancer.4,5
Autoimmune pancreatitis appears to be a disease of the elderly, as most patients are more than 50 years old at diagnosis. Twice as many men as women are affected.5 Many patients have no history of alcohol abuse or other traditional risk factors for chronic pancreatitis.
CLINICAL PRESENTATION: PAINLESS JAUNDICE, WEIGHT LOSS
The most common clinical presentation is obstructive jaundice with little or no abdominal pain. In one series,4 65% of patients presented with painless jaundice secondary to biliary obstruction. Obstructive acute pancreatitis can occur, due to inflammatory strictures of the main pancreatic duct.
Weight loss results from impaired digestion and decreased appetite. Autoimmune pancreatitis is complicated by pancreatic exocrine insufficiency in 88% of cases6 and by endocrine dysfunction in 67%.3
Many patients have extrapancreatic lesions such as sclerosing sialadenitis, retroperitoneal fibrosis, and autoimmune sclerosing cholangitis.7 The cholangiographic appearance of autoimmune sclerosing cholangitis may resemble that of primary sclerosing cholangitis or cholangiocarcinoma. Less common extrapancreatic findings include interstitial nephritis and mediastinal adenopathy. These extrapancreatic findings do not always coincide with pancreatic inflammation. The histopathologic findings in extrapancreatic lesions parallel those in the pancreas.8
The patient described at the beginning of this article had several of these features, including painless jaundice, weight loss, elevated alkaline phosphatase, and an inflammatory pancreatic mass.
DIAGNOSIS IS IMPROVING
Laboratory findings
Serum amylase and lipase are neither sensitive nor specific for autoimmune pancreatitis. Usually, their values are within normal limits or only mildly elevated.
A cholestatic pattern of elevation (elevated alkaline phosphatase and bilirubin, with normal or only slightly elevated alanine and aspartate aminotransferases) is found in patients with an inflammatory mass in the pancreatic head and in those with autoimmune sclerosing cholangitis. In one series,4 pancreatic enzymes were elevated in only 3 (13%) of 17 cases, while cholestasis was present in 16 (94%).
Gamma globulin, total IgG, and IgG4 are commonly elevated in autoimmune pancreatitis. Serum IgG4 is considered the most sensitive and specific marker and is elevated in 63% to 94% of patients with autoimmune pancreatitis.4,15–17 Several studies found the diagnostic accuracy, sensitivity, and specificity to be highest (> 90%) when a cut point of 135 mg/dL was used.17,18 A subsequent study 15 revealed a sensitivity of 76% and a specificity of 93% using the same cut point. Recall that the IgG4 level in our patient was 380 mg/dL.
Autoantibodies that are elevated in autoimmune pancreatitis include antilactoferrin antibodies and anticarbonic anhydrase II antibodies. 19 Both are “organ-specific”: the former are found in pancreatic acinar cells, and the latter are found in ductal cells. The sensitivity of both antibodies is greater than 50% in patients with autoimmune pancreatitis. However, they are not often measured, since testing for them is not widely available.20,21
Antinuclear antibody and rheumatoid factor are also associated with autoimmune pancreatitis but are not very specific.
Radiographic findings
The most common radiographic feature is diffuse enlargement of the entire pancreas. The appearance of the gland is often described as “sausage-like,” a feature best seen with CT and magnetic resonance imaging (MRI).
A well-defined capsule-like rim surrounding the pancreas is another common feature.23 This rim-enhancement is hypointense on T2 MRI, suggesting the presence of peripheral inflammation and fibrosis.
Calcifications and pseudocysts are rarely seen in autoimmune pancreatitis.
On ultrasonography, the involved pancreatic parenchyma appears hypoechoic, consistent with edema.
Endoscopic retrograde cholangiopancreatography
ERCP or magnetic resonance cholangiopancreatography may reveal segmental or diffuse narrowing of the main pancreatic duct.24,25 Bile-duct strictures may occur throughout the biliary tree.23
Autoimmune pancreatitis with biliary involvement must be distinguished from primary sclerosing cholangitis because the former responds to corticosteroid treatment. Cholangiographic features in primary sclerosing cholangitis include band-like strictures and a beaded or “pruned-tree” appearance, while autoimmune pancreatitis more commonly produces long strictures with prestenotic dilatation.26
ERCP allows temporary stents to be placed in obstructed segments of the biliary tree to open them up in the setting of acute cholangitis.
Biopsy guided by endoscopic ultrasonography
Some have proposed using endoscopic ultrasonography to guide biopsy in cases of suspected autoimmune pancreatitis.27,28
Fine-needle aspiration biopsy, guided by endoscopic ultrasonography, is frequently used to rule out adenocarcinoma. However, its yield for cancer is not perfect (about 70%–90%), so a negative biopsy does not rule out cancer. Further, autoimmune pancreatitis is rare, so a patient with a negative finding on fine-needle aspiration biopsy is still more likely to have cancer than autoimmune pancreatitis. In this case, the negative study should be combined with other information (eg, IgG4) to decide whether empiric treatment should be given.
Core biopsy, also guided by endoscopic ultrasonography, collects a greater amount of tissue for analysis and may allow the histologic diagnosis of autoimmune pancreatitis, but it carries a greater risk of bleeding. Also, its yield may be lower than initially thought. In one series, only 26% of ultrasonographically guided core samples from patients with confirmed autoimmune pancreatitis had diagnostic histologic features.29
New immunohistologic techniques are being developed to increase the yield from cytologic and tissue specimens.
Histopathologic findings
Histologic evaluation remains the gold standard for diagnosis. The histologic diagnosis can be made in patients who have any or all of the following three most common histologic features of autoimmune pancreatitis10,23,31–33:
- Parenchymal and often periductal lymphoplasmacytic infiltration, which is typically florid in intensity
- Storiform fibrosis
- Obliterative phlebitis.
The histologic findings in our patient included lymphoplasmacytic infiltration and obliterative phlebitis, which were essential to establishing the diagnosis. In a series of 53 patients, parenchymal inflammation with periductal lymphoplasmacytic accentuation was found in all of them.33
Biopsy of extrapancreatic sites, including the bile ducts and major duodenal papilla, may also facilitate the diagnosis.34,35 In a recent study,34 80% of autoimmune pancreatitis patients with pancreatic head involvement had significant numbers of IgG4-positive cells on biopsy of the major duodenal papilla. Biopsy of the periampullary duodenum may be a safer alternative to guided fine-needle aspiration or core biopsy.
In addition to lymphocytes, the inflammatory infiltrates in autoimmune pancreatitis may contain macrophages, mast cells, neutrophils, and eosinophils. Nonnecrotizing granulomas are occasionally seen, including periductal granulomas.
Fibrosis. Ductal luminal destruction can be seen in conjunction with fibrosis that thickens the duct wall and forms interlobular septa.33 Fibrosis may also affect the acinar tissue and produce profound lobular atrophy. In severe cases, the fibrotic changes can encompass large areas, with myofibroblasts arranged in a storiform pattern resembling an inflammatory pseudotumor.36
Phlebitis. The vascular changes in autoimmune pancreatitis have been underemphasized relative to the pancreatic parenchymal fibroinflammatory changes. Venulitis is seen mainly in small and medium-size pancreatic and peripancreatic veins. The inflammatory response and fibrosis disrupt the venous endothelium and often result in obliterative phlebitis.
However, Movat staining may not be available if an operative frozen section is being analyzed. In these cases, the venous lesions can be found by localizing the paired arteries, which are usually entirely normal and readily evident. If paired veins are not seen in this manner, a high level of suspicion should be raised for autoimmune pancreatitis with lymphocytic vein destruction.
CORTICOSTEROIDS ARE EFFECTIVE
Our patient’s jaundice temporarily resolved after his biliary bypass operation. If the diagnosis had been made earlier, he could have been treated with corticosteroids.
Corticosteroids have been used to treat autoimmune pancreatitis, with great success. (However, autoimmune pancreatitis occasional resolves spontaneously and stays in remission without corticosteroids.) A common regimen is oral prednisone 40 mg/day for 4 weeks and then tapered by 5 mg every 1 to 2 weeks. Patients who have a delayed response may receive long-term maintenance corticosteroid therapy (2.5–5 mg of oral prednisone).38–40
The radiographic and laboratory abnormalities typically resolve promptly with steroid therapy. A radiographic response is seen as early as 2 to 3 weeks, with normalization occurring in 4 to 6 weeks.40 Serum IgG4 levels decrease concurrently.38
Between 36% and 60% of patients with diabetes and autoimmune pancreatitis have better insulin secretion and glycemic control once corticosteroid therapy is started.3,6,38,40 Fifty percent of patients with exocrine insufficiency have functional improvement after corticosteroid therapy.6
Extrapancreatic lesions also improve with therapy.40,41 Obstructive jaundice may require endoscopic placement of a temporary biliary stent, but after a few weeks of steroid therapy the stent can usually be removed.
The decision to treat with corticosteroids is usually based on symptoms, imaging features (stricture or mass), a low suspicion of cancer (eg, negative biopsy), and an elevated IgG4. A histologic diagnosis of autoimmune pancreatitis is usually not available or required but may be sought through endoscopic ultrasonography-guided core biopsy or laparoscopic biopsy if the diagnosis is in doubt.
Another reasonable approach is an empiric trial of corticosteroids, reassessing the symptoms and repeating the imaging tests after 1 to 2 months. In fact, a response to corticosteroids is a component of most diagnostic criteria (Table 2).
Recurrence rates range from 6% to 32%.4,33,39,42,43 Patients who relapse after initial corticosteroid therapy may be treated again with prednisone in high doses (40 mg/day).38,41 Immunomodulatory therapy has been used successfully to treat relapsed disease in a single reported series: seven patients received either azathioprine (Imuran) 2 mg/kg daily or mycophenolate mofetil (Cell-Cept) 750 mg twice daily, and all remained in complete remission at a median follow-up of 6 months with no adverse events.44
In cases that fail to respond to corticosteroids, the diagnosis of autoimmune pancreatitis should be re-evaluated and surgery should be considered to look for cancer.
CASE CONTINUED
Our patient felt well at his 2-month follow-up visit. However, his serum alkaline phosphatase had increased to 649 U/L, and his serum IgG4 had increased to 980 mg/dL.
ERCP repeated 6 weeks later showed that the hilar stricture had completely resolved, and the intrahepatic strictures had markedly improved (Figure 6). His serum alkaline phosphatase level was now 73 U/L, and his serum IgG4 was 231 mg/dL.
Almost 2 years after starting corticosteroid therapy, the patient has remained in good control and the prednisone has been tapered off completely. His latest laboratory values are alkaline phosphatase 70 U/L and IgG4 46 mg/dL.
- Sarles H, Sarles JC, Muratore R, Guien C. Chronic inflammatory sclerosis of the pancreas—an autonomous pancreatic disease? Am J Dig Dis 1961; 6:688–698.
- Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci 1995; 40:1561–1568.
- Nishimori I, Tamakoshi A, Kawa S, et al; Research Committee on Intractable Pancreatic Diseases, the Ministry of Health and Welfare of Japan. Influence of steroid therapy on the course of diabetes mellitus in patients with autoimmune pancreatitis: findings from a nationwide survey in Japan. Pancreas 2006; 32:244–248.
- Kim KP, Kim M, Lee YJ, et al. Clinical characteristics of 17 cases of autoimmune chronic pancreatitis. Korean J Gastroenterol 2004; 43:112–119.
- Finkelberg DL, Sahani D, Deshpande V, Brugge WR. Autoimmune pancreatitis. N Engl J Med 2006; 355:2670–2676.
- Kamisawa T, Egawa N, Inokuma S, et al. Pancreatic endocrine and exocrine function and salivary gland function in autoimmune pancreatitis before and after steroid therapy. Pancreas 2003; 27:235–238.
- Kamisawa T, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Extrapancreatic lesions in autoimmune pancreatitis. J Clin Gastroenterol 2005; 39:904–907.
- Kamisawa T, Nakajima H, Egawa N, Funata N, Tsuruta K, Okamoto A. IgG4-related sclerosing disease incorporating sclerosing pancreatitis, cholangitis, sialadenitis and retroperitoneal fibrosis with lymphadenopathy. Pancreatology 2006; 6:132–137.
- Kwon S, Kim MH, Choi EK. The diagnostic criteria for autoimmune chronic pancreatitis: it is time to make a consensus. Pancreas 2007; 34:279–286.
- Chari ST. Diagnosis of autoimmune pancreatitis using its five cardinal features: introducing The Mayo Clinic’s HISORt criteria. J Gastroenterol 2007; 42( suppl 18):39–41.
- Chari ST, Smyrk TC, Levy MJ, et al. Diagnosis of autoimmune pancreatitis: The Mayo Clinic experience. Clin Gastroenterol Hepatol 2006; 4:1010–1016.
- Kim K-P, Kim M-H, Kim JC, Lee SS, Seo DW, Lee SK. Diagnostic criteria for autoimmune chronic pancreatitis revisited. World J Gastroenterol 2006; 12:2487–2496.
- Okazaki K, Kawa S, Kamisawa T. Clinical diagnostic criteria of autoimmune pancreatitis: revised proposal. J Gastroenterol 2006; 41:626–631.
- Nishimori I, Onishi S, Otsuki M. Review of diagnostic criteria for autoimmune pancreatitis; for establishment of international criteria. Clin J Gastroenterol 2008; 1:7–17.
- Ghazale A, Chari ST, Smyrk TC, et al Value of serum IgG4 in the diagnosis of autoimmune pancreatitis and in distinguishing it from pancreatic cancer. Am J Gastroenterol 2007; 102:1646–1653.
- Hirano K, Kawabe T, Yamamoto N, et al. Serum IgG4 concentrations in pancreatic and biliary diseases. Clin Chim Acta 2006; 367:181–184.
- Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001; 344:732–738.
- Kawa S, Hamano H. Clinical features of autoimmune pancreatitis. J Gastroenterol 2007; 42(suppl 18):9–14.
- Okazaki K, Uchida K, Ohana M, et al. Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology 2000; 118:573–581.
- Frulloni L, Bovo P, Brunelli S, et al. Elevated serum levels of antibodies to carbonic anhydrase I and II in patients with chronic pancreatitis. Pancreas 2000; 20:382–388.
- Nishimori I, Miyaji E, Morimoto K, Nagao K, Kamada M, Onishi S. Serum antibodies to carbonic anhydrase IV in patients with autoimmune pancreatitis. Gut 2005; 54:274–281.
- Irie H, Honda H, Baba S, et al. Autoimmune pancreatitis: CT and MR characteristics. AJR Am J Roentgenol 1998; 170:1323–1327.
- Sahani DV, Kalva SP, Farrell J, et al. Autoimmune pancreatitis: imaging features. Radiology 2004; 233:345–352.
- Horiuchi A, Kawa S, Hamano H, Hayama M, Ota H, Kiyosawa K. ERCP features in 27 patients with autoimmune pancreatitis. Gastrointest Endosc 2002; 55:494–499.
- Kamisawa T, Chen PY, Tu Y, et al. MRCP and MRI findings in 9 patients with autoimmune pancreatitis. World J Gastroenterol 2006; 12:2919–2922.
- Nakazawa T, Ohara H, Sano H, et al. Cholangiography can discriminate sclerosing cholangitis with autoimmune pancreatitis from primary sclerosing cholangitis. Gastrointest Endosc 2004; 60:937–944.
- Farrell JJ, Garber J, Sahani D, Brugge WR. EUS findings in patients with autoimmune pancreatitis. Gastrointest Endosc 2004; 60:927–936.
- Levy MJ, Reddy RP, Wiersema MJ, et al. EUS-guided trucut biopsy in establishing autoimmune pancreatitis as the cause of obstructive jaundice. Gastrointest Endosc 2005; 61:467–472.
- Bang SJ, Kim MH, Kim do H, et al. Is pancreatic core biopsy sufficient to diagnose autoimmune chronic pancreatitis? Pancreas 2008; 36:84–89.
- Scully KA, Li SC, Hebert JC, Trainer TD. The characteristic appearance of non-alcoholic duct destructive chronic pancreatitis: a report of 2 cases. Arch Pathol Lab Med 2000; 124:1535–1538.
- Chu KE, Papouchado BG, Lane Z, Bronner MP. The role of Movat pentachrome stain and immunoglobulin G4 immunostaining in the diagnosis of autoimmune pancreatitis. Mod Pathol 2009; 22:351–358.
- Ectors N, Maillet B, Aerts R, et al. Non-alcoholic duct destructive chronic pancreatitis. Gut 1997; 41:263–268.
- Zamboni G, Luttges J, Capelli P, et al. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch 2004; 445:552–563.
- Hamano H, Kawa S, Uehara T, et al. Immunoglobulin G4-related lymphoplasmacytic sclerosing cholangitis that mimics infiltrating hilar cholangiocarcinoma: part of a spectrum of autoimmune pancreatitis? Gastrointest Endosc 2005; 62:152–157.
- Kamisawa T, Tu Y, Egawa N, Tsuruta K, Okamoto A. A new diagnostic endoscopic tool for autoimmune pancreatitis. Gastrointest Endosc 2008; 68:358–361.
- Notohara K, Burgart LJ, Yadav D, Chari S, Smyrk TC. Idiopathic chronic pancreatitis with periductal lymphoplasmacytic infiltration: clinicopathologic features of 35 cases. Am J Surg Pathol 2003; 27:1119–1127.
- Esposito I, Bergmann F, Penzel R, et al. Oligoclonal T-cell populations in an inflammatory pseudotumor of the pancreas possibly related to autoimmune pancreatitis: an immunohistochemical and molecular analysis. Virchows Arch 2004; 444:119–126.
- Kamisawa T, Okamoto A, Wakabayashi T, Watanabe H, Sawabu N. Appropriate steroid therapy for autoimmune pancreatitis based on long-term outcome. Scand J Gastroenterol 2008; 43:609–613.
- Hirano K, Tada M, Isayama H, et al. Long-term prognosis of autoimmune pancreatitis with and without corticosteroid treatment. Gut 2007; 56:1719–1724.
- Kamisawa T, Yoshiike M, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Treating patients with autoimmune pancreatitis: results from a long-term follow-up study. Pancreatology 2005; 5:234–38.
- Kamisawa T, Okamoto A. Prognosis of autoimmune pancreatitis. J Gastroenterol 2007; 42(suppl 18):59–62.
- Takayama M, Hamano H, Ochi Y, et al. Recurrent attacks of autoimmune pancreatitis result in pancreatic stone formation. Am J Gastroenterol 2004; 99:932–937.
- Wakabayashi T, Kawaura Y, Satomura Y, Watanabe H, Motoo Y, Sawabu N. Long-term prognosis of duct-narrowing chronic pancreatitis: strategy for steroid treatment. Pancreas 2005; 30:31–39.
- Ghazale A, Chari ST. Optimising corticosteroid treatment for autoimmune pancreatitis. Gut 2007; 56:1650–1652.
- Sarles H, Sarles JC, Muratore R, Guien C. Chronic inflammatory sclerosis of the pancreas—an autonomous pancreatic disease? Am J Dig Dis 1961; 6:688–698.
- Yoshida K, Toki F, Takeuchi T, Watanabe S, Shiratori K, Hayashi N. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci 1995; 40:1561–1568.
- Nishimori I, Tamakoshi A, Kawa S, et al; Research Committee on Intractable Pancreatic Diseases, the Ministry of Health and Welfare of Japan. Influence of steroid therapy on the course of diabetes mellitus in patients with autoimmune pancreatitis: findings from a nationwide survey in Japan. Pancreas 2006; 32:244–248.
- Kim KP, Kim M, Lee YJ, et al. Clinical characteristics of 17 cases of autoimmune chronic pancreatitis. Korean J Gastroenterol 2004; 43:112–119.
- Finkelberg DL, Sahani D, Deshpande V, Brugge WR. Autoimmune pancreatitis. N Engl J Med 2006; 355:2670–2676.
- Kamisawa T, Egawa N, Inokuma S, et al. Pancreatic endocrine and exocrine function and salivary gland function in autoimmune pancreatitis before and after steroid therapy. Pancreas 2003; 27:235–238.
- Kamisawa T, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Extrapancreatic lesions in autoimmune pancreatitis. J Clin Gastroenterol 2005; 39:904–907.
- Kamisawa T, Nakajima H, Egawa N, Funata N, Tsuruta K, Okamoto A. IgG4-related sclerosing disease incorporating sclerosing pancreatitis, cholangitis, sialadenitis and retroperitoneal fibrosis with lymphadenopathy. Pancreatology 2006; 6:132–137.
- Kwon S, Kim MH, Choi EK. The diagnostic criteria for autoimmune chronic pancreatitis: it is time to make a consensus. Pancreas 2007; 34:279–286.
- Chari ST. Diagnosis of autoimmune pancreatitis using its five cardinal features: introducing The Mayo Clinic’s HISORt criteria. J Gastroenterol 2007; 42( suppl 18):39–41.
- Chari ST, Smyrk TC, Levy MJ, et al. Diagnosis of autoimmune pancreatitis: The Mayo Clinic experience. Clin Gastroenterol Hepatol 2006; 4:1010–1016.
- Kim K-P, Kim M-H, Kim JC, Lee SS, Seo DW, Lee SK. Diagnostic criteria for autoimmune chronic pancreatitis revisited. World J Gastroenterol 2006; 12:2487–2496.
- Okazaki K, Kawa S, Kamisawa T. Clinical diagnostic criteria of autoimmune pancreatitis: revised proposal. J Gastroenterol 2006; 41:626–631.
- Nishimori I, Onishi S, Otsuki M. Review of diagnostic criteria for autoimmune pancreatitis; for establishment of international criteria. Clin J Gastroenterol 2008; 1:7–17.
- Ghazale A, Chari ST, Smyrk TC, et al Value of serum IgG4 in the diagnosis of autoimmune pancreatitis and in distinguishing it from pancreatic cancer. Am J Gastroenterol 2007; 102:1646–1653.
- Hirano K, Kawabe T, Yamamoto N, et al. Serum IgG4 concentrations in pancreatic and biliary diseases. Clin Chim Acta 2006; 367:181–184.
- Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001; 344:732–738.
- Kawa S, Hamano H. Clinical features of autoimmune pancreatitis. J Gastroenterol 2007; 42(suppl 18):9–14.
- Okazaki K, Uchida K, Ohana M, et al. Autoimmune-related pancreatitis is associated with autoantibodies and a Th1/Th2-type cellular immune response. Gastroenterology 2000; 118:573–581.
- Frulloni L, Bovo P, Brunelli S, et al. Elevated serum levels of antibodies to carbonic anhydrase I and II in patients with chronic pancreatitis. Pancreas 2000; 20:382–388.
- Nishimori I, Miyaji E, Morimoto K, Nagao K, Kamada M, Onishi S. Serum antibodies to carbonic anhydrase IV in patients with autoimmune pancreatitis. Gut 2005; 54:274–281.
- Irie H, Honda H, Baba S, et al. Autoimmune pancreatitis: CT and MR characteristics. AJR Am J Roentgenol 1998; 170:1323–1327.
- Sahani DV, Kalva SP, Farrell J, et al. Autoimmune pancreatitis: imaging features. Radiology 2004; 233:345–352.
- Horiuchi A, Kawa S, Hamano H, Hayama M, Ota H, Kiyosawa K. ERCP features in 27 patients with autoimmune pancreatitis. Gastrointest Endosc 2002; 55:494–499.
- Kamisawa T, Chen PY, Tu Y, et al. MRCP and MRI findings in 9 patients with autoimmune pancreatitis. World J Gastroenterol 2006; 12:2919–2922.
- Nakazawa T, Ohara H, Sano H, et al. Cholangiography can discriminate sclerosing cholangitis with autoimmune pancreatitis from primary sclerosing cholangitis. Gastrointest Endosc 2004; 60:937–944.
- Farrell JJ, Garber J, Sahani D, Brugge WR. EUS findings in patients with autoimmune pancreatitis. Gastrointest Endosc 2004; 60:927–936.
- Levy MJ, Reddy RP, Wiersema MJ, et al. EUS-guided trucut biopsy in establishing autoimmune pancreatitis as the cause of obstructive jaundice. Gastrointest Endosc 2005; 61:467–472.
- Bang SJ, Kim MH, Kim do H, et al. Is pancreatic core biopsy sufficient to diagnose autoimmune chronic pancreatitis? Pancreas 2008; 36:84–89.
- Scully KA, Li SC, Hebert JC, Trainer TD. The characteristic appearance of non-alcoholic duct destructive chronic pancreatitis: a report of 2 cases. Arch Pathol Lab Med 2000; 124:1535–1538.
- Chu KE, Papouchado BG, Lane Z, Bronner MP. The role of Movat pentachrome stain and immunoglobulin G4 immunostaining in the diagnosis of autoimmune pancreatitis. Mod Pathol 2009; 22:351–358.
- Ectors N, Maillet B, Aerts R, et al. Non-alcoholic duct destructive chronic pancreatitis. Gut 1997; 41:263–268.
- Zamboni G, Luttges J, Capelli P, et al. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch 2004; 445:552–563.
- Hamano H, Kawa S, Uehara T, et al. Immunoglobulin G4-related lymphoplasmacytic sclerosing cholangitis that mimics infiltrating hilar cholangiocarcinoma: part of a spectrum of autoimmune pancreatitis? Gastrointest Endosc 2005; 62:152–157.
- Kamisawa T, Tu Y, Egawa N, Tsuruta K, Okamoto A. A new diagnostic endoscopic tool for autoimmune pancreatitis. Gastrointest Endosc 2008; 68:358–361.
- Notohara K, Burgart LJ, Yadav D, Chari S, Smyrk TC. Idiopathic chronic pancreatitis with periductal lymphoplasmacytic infiltration: clinicopathologic features of 35 cases. Am J Surg Pathol 2003; 27:1119–1127.
- Esposito I, Bergmann F, Penzel R, et al. Oligoclonal T-cell populations in an inflammatory pseudotumor of the pancreas possibly related to autoimmune pancreatitis: an immunohistochemical and molecular analysis. Virchows Arch 2004; 444:119–126.
- Kamisawa T, Okamoto A, Wakabayashi T, Watanabe H, Sawabu N. Appropriate steroid therapy for autoimmune pancreatitis based on long-term outcome. Scand J Gastroenterol 2008; 43:609–613.
- Hirano K, Tada M, Isayama H, et al. Long-term prognosis of autoimmune pancreatitis with and without corticosteroid treatment. Gut 2007; 56:1719–1724.
- Kamisawa T, Yoshiike M, Egawa N, Nakajima H, Tsuruta K, Okamoto A. Treating patients with autoimmune pancreatitis: results from a long-term follow-up study. Pancreatology 2005; 5:234–38.
- Kamisawa T, Okamoto A. Prognosis of autoimmune pancreatitis. J Gastroenterol 2007; 42(suppl 18):59–62.
- Takayama M, Hamano H, Ochi Y, et al. Recurrent attacks of autoimmune pancreatitis result in pancreatic stone formation. Am J Gastroenterol 2004; 99:932–937.
- Wakabayashi T, Kawaura Y, Satomura Y, Watanabe H, Motoo Y, Sawabu N. Long-term prognosis of duct-narrowing chronic pancreatitis: strategy for steroid treatment. Pancreas 2005; 30:31–39.
- Ghazale A, Chari ST. Optimising corticosteroid treatment for autoimmune pancreatitis. Gut 2007; 56:1650–1652.
KEY POINTS
- Hallmark features of autoimmune pancreatitis include an elevated serum immunoglobulin G4 level, focal or diffuse pancreatic enlargement on imaging, and dense lymphoplasmacytic infiltrates on histologic study.
- The disease can be associated with extrapancreatic manifestations, including sclerosing cholangitis, sialadenitis and retroperitoneal fibrosis.
- Autoimmune pancreatitis responds dramatically to corticosteroid treatment.