User login
July 2021 - What's the diagnosis?
Answer: Erythropoietic protoporphyria.Figure B demonstrated massive cholestasis with brown deposits that represented protoporphyrin precipitates, which plugged the bile ducts and led to a cholestatic pattern of liver injury. Under polarized light, protoporhyrin precipitates produced Maltese crosses (Figure C), which are pathognomonic of erythropoietic protoporphyria (EPP). Porphyria is a rare group of inherited heme biosynthesis disorders. EPP is an uncommon type of porphyria and is secondary to a ferrochelatase (FECH) gene mutation, which results in deficient activity of the mitochondrial enzyme FECH.1
FECH catalyzes chelation of iron into proptoporphyrin IX to form heme. The inability of protoporphyrins to be transformed into heme inhibits hepatic elimination and results in hepatocyte accumulation of protoporphyrins, leading to protoporphyrin precipitation in bile canaliculi. Painful photosensitivity (Figure A) is the most common manifestation of EPP, beginning in childhood.2 Only a small proportion of patients with EPP develop liver dysfunction but the consequences can be severe.2 Therefore, therapeutic decisions are based on limited published experience without randomized, controlled data.2 One treatment method is to attempt to remove protoporphyrins from the blood via therapeutic plasma exchange.2Our patient underwent one session of therapeutic plasma exchange; however, after this initial course of treatment, the patient’s goals of care changed and she elected to enroll in hospice. Patients with severe liver dysfunction as a result of EPP require consideration of liver transplantation in the setting of fulminant hepatic failure. Liver transplantation does not cure EPP; the graft is at risk for similar EPP-related changes.1 Only bone marrow transplantation can correct the underlying enzymatic defect in FECH.1 Although physicians are often taught “common things are common,” this case highlights a rare complication of a rare disease such as porphyria is an often forgotten or missed condition. Vigilance should be kept for other rare conditions, especially ones with curative treatments or fatal consequences. In an era where the role of liver biopsy is often questioned in favor of prediction models or noninvasive testing, we must have a low threshold to safely perform a liver biopsy when the diagnosis is unclear or a patient is deteriorating.
The quiz authors disclosed no conflicts of interest.
References
1. Windon AL et al. Am J Transplant. 2018 Mar;18(3):745-9.
2. Pagano MB et al. J Clin Apher. 2012;27(6):336-41.
Answer: Erythropoietic protoporphyria.Figure B demonstrated massive cholestasis with brown deposits that represented protoporphyrin precipitates, which plugged the bile ducts and led to a cholestatic pattern of liver injury. Under polarized light, protoporhyrin precipitates produced Maltese crosses (Figure C), which are pathognomonic of erythropoietic protoporphyria (EPP). Porphyria is a rare group of inherited heme biosynthesis disorders. EPP is an uncommon type of porphyria and is secondary to a ferrochelatase (FECH) gene mutation, which results in deficient activity of the mitochondrial enzyme FECH.1
FECH catalyzes chelation of iron into proptoporphyrin IX to form heme. The inability of protoporphyrins to be transformed into heme inhibits hepatic elimination and results in hepatocyte accumulation of protoporphyrins, leading to protoporphyrin precipitation in bile canaliculi. Painful photosensitivity (Figure A) is the most common manifestation of EPP, beginning in childhood.2 Only a small proportion of patients with EPP develop liver dysfunction but the consequences can be severe.2 Therefore, therapeutic decisions are based on limited published experience without randomized, controlled data.2 One treatment method is to attempt to remove protoporphyrins from the blood via therapeutic plasma exchange.2Our patient underwent one session of therapeutic plasma exchange; however, after this initial course of treatment, the patient’s goals of care changed and she elected to enroll in hospice. Patients with severe liver dysfunction as a result of EPP require consideration of liver transplantation in the setting of fulminant hepatic failure. Liver transplantation does not cure EPP; the graft is at risk for similar EPP-related changes.1 Only bone marrow transplantation can correct the underlying enzymatic defect in FECH.1 Although physicians are often taught “common things are common,” this case highlights a rare complication of a rare disease such as porphyria is an often forgotten or missed condition. Vigilance should be kept for other rare conditions, especially ones with curative treatments or fatal consequences. In an era where the role of liver biopsy is often questioned in favor of prediction models or noninvasive testing, we must have a low threshold to safely perform a liver biopsy when the diagnosis is unclear or a patient is deteriorating.
The quiz authors disclosed no conflicts of interest.
References
1. Windon AL et al. Am J Transplant. 2018 Mar;18(3):745-9.
2. Pagano MB et al. J Clin Apher. 2012;27(6):336-41.
Answer: Erythropoietic protoporphyria.Figure B demonstrated massive cholestasis with brown deposits that represented protoporphyrin precipitates, which plugged the bile ducts and led to a cholestatic pattern of liver injury. Under polarized light, protoporhyrin precipitates produced Maltese crosses (Figure C), which are pathognomonic of erythropoietic protoporphyria (EPP). Porphyria is a rare group of inherited heme biosynthesis disorders. EPP is an uncommon type of porphyria and is secondary to a ferrochelatase (FECH) gene mutation, which results in deficient activity of the mitochondrial enzyme FECH.1
FECH catalyzes chelation of iron into proptoporphyrin IX to form heme. The inability of protoporphyrins to be transformed into heme inhibits hepatic elimination and results in hepatocyte accumulation of protoporphyrins, leading to protoporphyrin precipitation in bile canaliculi. Painful photosensitivity (Figure A) is the most common manifestation of EPP, beginning in childhood.2 Only a small proportion of patients with EPP develop liver dysfunction but the consequences can be severe.2 Therefore, therapeutic decisions are based on limited published experience without randomized, controlled data.2 One treatment method is to attempt to remove protoporphyrins from the blood via therapeutic plasma exchange.2Our patient underwent one session of therapeutic plasma exchange; however, after this initial course of treatment, the patient’s goals of care changed and she elected to enroll in hospice. Patients with severe liver dysfunction as a result of EPP require consideration of liver transplantation in the setting of fulminant hepatic failure. Liver transplantation does not cure EPP; the graft is at risk for similar EPP-related changes.1 Only bone marrow transplantation can correct the underlying enzymatic defect in FECH.1 Although physicians are often taught “common things are common,” this case highlights a rare complication of a rare disease such as porphyria is an often forgotten or missed condition. Vigilance should be kept for other rare conditions, especially ones with curative treatments or fatal consequences. In an era where the role of liver biopsy is often questioned in favor of prediction models or noninvasive testing, we must have a low threshold to safely perform a liver biopsy when the diagnosis is unclear or a patient is deteriorating.
The quiz authors disclosed no conflicts of interest.
References
1. Windon AL et al. Am J Transplant. 2018 Mar;18(3):745-9.
2. Pagano MB et al. J Clin Apher. 2012;27(6):336-41.
A 66-year-old White woman with tetralogy of Fallot status after remote pulmonic valve surgery, hypothyroidism, and previous cholecystectomy presented to her primary care provider with 2 days of constant, dull, right upper quadrant pain with nausea but without fever, association with meals, or association with defecation. Her home medications included low-dose aspirin and levothyroxine. Her physical examination revealed normal vital signs, a body mass index of 29 kg/m2, right upper quadrant tenderness to palpation without peritoneal signs, and normal bowel sounds. The remainder of her examination was normal.
The patient underwent an exhaustive evaluation beginning with laboratory tests, which revealed a normal complete blood count, basic metabolic panel, lipase, international normalized ratio, and urinalysis. Her liver function tests results showed aspartate aminotransferase 118 international IU/L, alanine aminotransferase 117 IU/L, alkaline phosphatase 147 IU/L, and total bilirubin 17.6 mg/dL, with a direct bilirubin of 11.9 mg/dL.
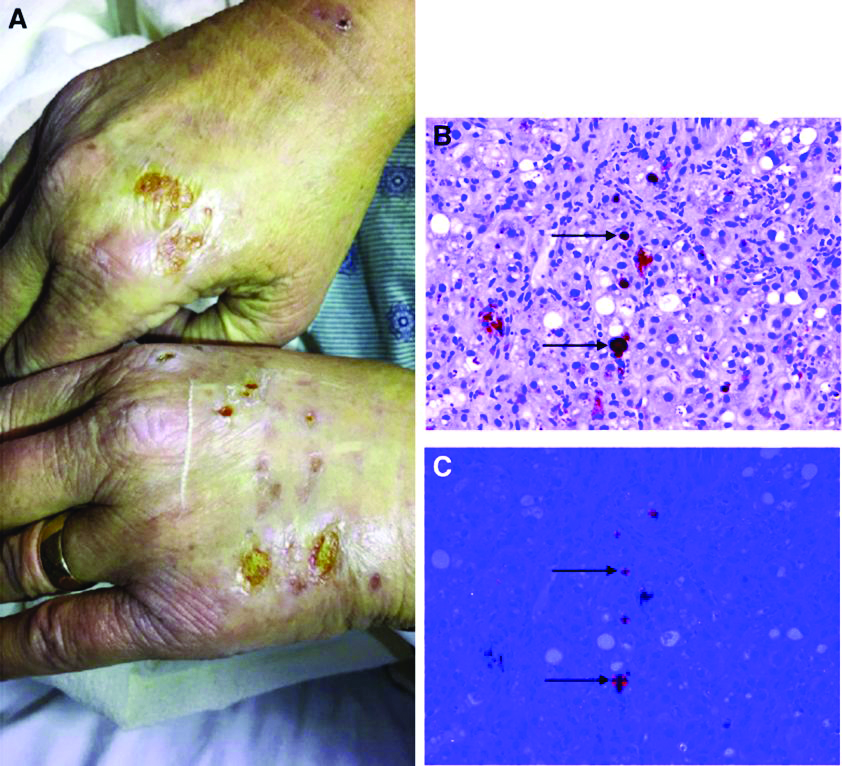
Her liver function tests were last checked 18 months prior and were normal. A liver ultrasound examination revealed cirrhotic morphology without ascites or hepatic or portal vein thrombosis. A magnetic resonance imaging study of the liver revealed morphologic changes of hepatic cirrhosis without portal hypertension, biliary dilation, or stricturing. Additionally, hepatitis A IgM, hepatitis B surface antigen, hepatitis B core IgM and IgG, hepatitis C antibody, ceruloplasmin, antinuclear antibody, anti-smooth muscle antibody, anti-liver-kidney-microsomal antibody, quantitative immunoglobulins, antimitochondrial antibody, alpha-1 antitrypsin phenotype, phosphatidylethanolamine, serum protein electrophoresis, and alpha fetoprotein were reassuring. Later, the patient reported sensitivity to the sun, described as a "sun allergy" with irritation on her hands (Figure A). Mentation remained normal; however, given progressive worsening hepatic function evidenced by international normalized ratio of 1.7 and bilirubin of 27.6 mg/dL, the patient was urgently admitted for expedited portal manometry with transjugular liver biopsy. The hepatic venous pressure gradient was 23 mm Hg. The liver biopsy images are shown in Figure B, C.