User login
Avoiding common drug−drug interactions
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight

hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
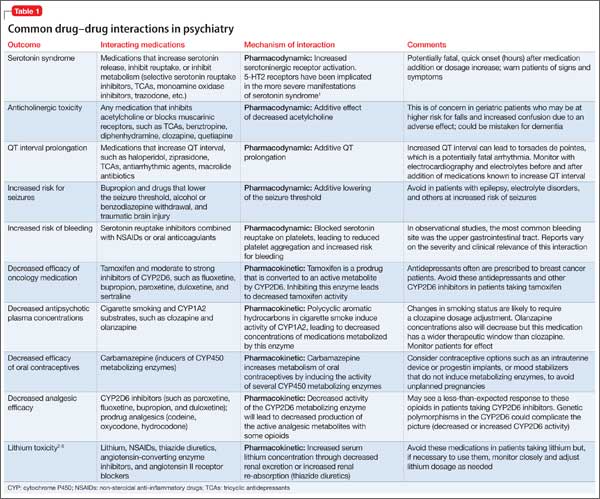
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight
hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight
hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
Glutamatergic therapies

Do glutamatergic drugs have a role in treating depression?
Mrs. S, age 46, has been struggling to manage depression for 7 years. She completed adequate trials of several selective serotonin reuptake inhibitors and bupropion. Currently, she is taking duloxetine, 60 mg/d, and aripiprazole, 5 mg/d.
At her most recent clinic visit, Mrs. S reports that she is doing “OK,” but that she still feels sad and disengaged most days of the week. She wants to know more about ketamine for treating depression after reading about it on the Internet and hearing it mentioned in a support group she attends. She asks if you think it would work for her, and gives you with a copy of an article about its use in patients with treatment-resistant depression. Mrs. S has no other health conditions and takes a daily vitamin D and calcium supplement.
The monoamine hypothesis of depression postulates that symptoms originate from underactivity of monoamines, such as serotonin, norepinephrine, and dopamine, in the brain. This hypothesis was formulated in the 1960s after researchers observed that monoamine oxidase inhibitors and tricyclic antidepressants relieved depressive symptoms; both were known to increase monoamine concentrations in the synaptic cleft.1

Regrettably, these medications do not adequately relieve depressive symptoms for many people. In fact, symptom remission occurs in only one-third of treated patients.2 This low remission rate reflects a lack of understanding of the pathophysiology of depression, and the need for drugs with unique mechanisms of action.
One of the newest drug targets shown to be relevant in psychiatric illness is the
glutamatergic system. Glutamate is the predominant excitatory neurotransmitter in the CNS, and it is responsible for many key functions, including synaptic plasticity, learning, memory, and locomotion.3 Normally, the glutamatergic system tightly regulates the amount of glutamate in the neuronal synapse via receptors on presynaptic and postsynaptic neurons, as well as on glial cells (Figure). When this equilibrium is disrupted in stressful situations, such as ischemia, trauma, or seizures, excess glutamate is released into the synapse. The resulting glutamatergic hyperactivity can lead to neurotoxicity and cell death when neuronal receptors are activated for an extended period. 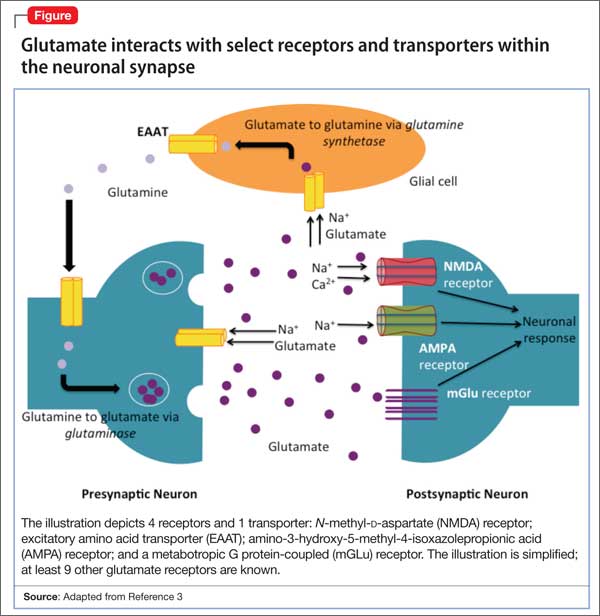
A key component of the glutamatergic system that is responsible for removing excess glutamate from the synapse is membrane-bound transporters, which are similar to serotonin and norepinephrine transporters. These excitatory amino acid transporters (EAATs) are important because glutamate metabolism does not occur within the synapse and EAATS are responsible for removing most of the glutamate from the synapse into glial cells.3
The network of receptors within the synapse that are activated by glutamate is extensive and complex. There are at least 11 glutamate-responsive receptors: 3 are ionotropic action channels, and the remaining 8 are metabotropic G protein-coupled receptors. Previous studies have shown regional changes in glutamate receptors, as well as elevated levels of glutamate, in the brains of patients with major depressive disorder (MDD).4
Ketamine. The ionotropic receptor N-methyl-d-aspartate (NMDA) is one of the most studied glutamate receptors. Pharmacologically, ketamine is a noncompetitive NMDA receptor antagonist that also activates the amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, which is another subtype of ionotropic glutamate receptors. In open-label clinical trials, ketamine has demonstrated rapid antidepressant action in patients with treatment-resistant MDD.4,5
Recently, Murrough et al6 performed the first randomized, psychoactive controlled trial using a single IV infusion of ketamine dosed below anesthesia ranges (0.5 mg/kg), or midazolam (0.045 mg/kg), in patients with treatment-resistant depression who had been antidepressant-free for at least 4 weeks. They found that 24 hours after medication administration, the likelihood of response to ketamine was significantly higher than the response to midazolam (OR: 2.18; 95% CI: 1.21 to 4.14), with a response rate of 64% in the ketamine group and 28% in the midazolam group.6
Psychotropic side effects, such as hallucinations, are a major concern with ketamine tolerability and abuse potential. This is largely because of ketamine’s antagonism of the NMDA receptor, which is a property shared with other abused drugs such as phencyclidine (PCP) and dextromethorphan. In the Murrough et al6 study, there were no reported cases of paranoia or hallucinations, but dissociative symptoms were relatively common (17%).
Although the results in this trial appear encouraging, there are several limitations to using ketamine to treat MDD, especially in an ambulatory setting. Concerns include ketamine’s IV administration, potential for abuse, long-term efficacy, and side-effect profile—particularly psychotic symptoms and hemodynamic changes. An ideal compound would have the rapid efficacy of ketamine, but with a safer side-effect profile, easier administration, and less potential for abuse.
Riluzole also acts on the glutamatergic system, but has not shown antidepressant efficacy as consistently as ketamine. Riluzole is FDA-approved for treating amyotrophic lateral sclerosis.5 Pharmacologically, riluzole is a glutamatergic modulator that increases glutamate reuptake into glial cells, decreases glutamate release, and increases AMPA trafficking. In open-label studies riluzole has shown efficacy in reducing depressive symptoms.4,5 However, when compared with placebo as a means of sustaining treatment response after a 1-time dose of ketamine, riluzole showed was no significant improvement in time to depressive relapse.7
Acamprosate, often used for treating alcohol abuse, is another a drug with glutamatergic activity that has been studied for possible use as an antidepressant.5
A review by Lapidus et al5 has a more extensive listing of current medications and investigational compounds that modulate glutamate transmission, and are of interest for their possible antidepressant activity. Given the relatively new “glutamatergic hypothesis” of depression, it is exciting that so many current and novel glutamatergic drug therapies are being evaluated.
Future of ketamine treatment
Glutamate has been shown to play an important part in the pathophysiology of depression. The rapid antidepressant efficacy of ketamine provides evidence that future medications with glutamate-modulating activity could be useful for patients who struggle to achieve symptom relief using available antidepressants. Several limitations exist regarding ketamine use, and more work in this important therapeutic area needs to be done. This last point is important to remember when speaking with patients such as Mrs. S. Although it is understandable for her to be excited about novel treatment options such as ketamine, stress to her that treating depression with ketamine at this time is strictly investigational, and that the drug needs to be thoroughly evaluated for safety and efficacy before it can be prescribed for this indication.
CASE CONTINUED
Mrs. S realizes that ketamine may not be the best next step for her, and she agrees to explore other approaches to treat her residual depressive symptoms.
Related Resources
• Machado-Vieira R, Ibrahim L, Henter ID, et al. Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol Biochem Behav. 2012;100(4):678-687.
• Mathews DC, Henter ID, Zarate CA. Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs. 2012;72(10):1313-1333.
Drug Brand Names
Acamprosate • Campral Duloxetine • Cymbalta
Aripiprazole • Abilify Ketamine • Ketalar
Bupropion • Wellbutrin, Zyban Riluzole • Rilutek
Disclosures
The authors report no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products.
1. Niciu MJ, Ionescu DF, Richards EM, et al. Glutamate and its receptors in the pathophysiology and treatment of major depressive disorder. J Neural Transm. 2014;121(8):907-924.
2. Gaynes BN, Dusetzina SB, Ellis AR, et al. Treating depression after initial treatment failure: directly comparing switch and augmenting strategies in STAR*D. J Clin Psychopharmacol. 2012;32(1):114-119.
3. Curry SC, Mills KC, Ruha A, et al. Neurotransmitters and neuromodulators. In: Nelson LS, Lewin NA, Howland MA, et al, eds. Goldfrank’s toxicologic emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:189-220.
4. Zarate C Jr, Machado-Vieira R, Henter I, et al. Glutamatergic modulators: the future of treating mood disorders? Harv Rev Psychiatry. 2010;18(5):293-303.
5. Lapidus KA, Soleimani L, Murrough JW. Novel glutamatergic drugs for the treatment of mood disorders. Neuropsychiatr Dis Treat. 2013;9:1101-1112.
6. Murrough JW, Iosifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
7. Ibrahim L, Diazgranados N, Franco-Chaves J, et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology. 2012;37(6):1526-1533.
Mrs. S, age 46, has been struggling to manage depression for 7 years. She completed adequate trials of several selective serotonin reuptake inhibitors and bupropion. Currently, she is taking duloxetine, 60 mg/d, and aripiprazole, 5 mg/d.
At her most recent clinic visit, Mrs. S reports that she is doing “OK,” but that she still feels sad and disengaged most days of the week. She wants to know more about ketamine for treating depression after reading about it on the Internet and hearing it mentioned in a support group she attends. She asks if you think it would work for her, and gives you with a copy of an article about its use in patients with treatment-resistant depression. Mrs. S has no other health conditions and takes a daily vitamin D and calcium supplement.
The monoamine hypothesis of depression postulates that symptoms originate from underactivity of monoamines, such as serotonin, norepinephrine, and dopamine, in the brain. This hypothesis was formulated in the 1960s after researchers observed that monoamine oxidase inhibitors and tricyclic antidepressants relieved depressive symptoms; both were known to increase monoamine concentrations in the synaptic cleft.1
Regrettably, these medications do not adequately relieve depressive symptoms for many people. In fact, symptom remission occurs in only one-third of treated patients.2 This low remission rate reflects a lack of understanding of the pathophysiology of depression, and the need for drugs with unique mechanisms of action.
One of the newest drug targets shown to be relevant in psychiatric illness is the
glutamatergic system. Glutamate is the predominant excitatory neurotransmitter in the CNS, and it is responsible for many key functions, including synaptic plasticity, learning, memory, and locomotion.3 Normally, the glutamatergic system tightly regulates the amount of glutamate in the neuronal synapse via receptors on presynaptic and postsynaptic neurons, as well as on glial cells (Figure). When this equilibrium is disrupted in stressful situations, such as ischemia, trauma, or seizures, excess glutamate is released into the synapse. The resulting glutamatergic hyperactivity can lead to neurotoxicity and cell death when neuronal receptors are activated for an extended period.
A key component of the glutamatergic system that is responsible for removing excess glutamate from the synapse is membrane-bound transporters, which are similar to serotonin and norepinephrine transporters. These excitatory amino acid transporters (EAATs) are important because glutamate metabolism does not occur within the synapse and EAATS are responsible for removing most of the glutamate from the synapse into glial cells.3
The network of receptors within the synapse that are activated by glutamate is extensive and complex. There are at least 11 glutamate-responsive receptors: 3 are ionotropic action channels, and the remaining 8 are metabotropic G protein-coupled receptors. Previous studies have shown regional changes in glutamate receptors, as well as elevated levels of glutamate, in the brains of patients with major depressive disorder (MDD).4
Ketamine. The ionotropic receptor N-methyl-d-aspartate (NMDA) is one of the most studied glutamate receptors. Pharmacologically, ketamine is a noncompetitive NMDA receptor antagonist that also activates the amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, which is another subtype of ionotropic glutamate receptors. In open-label clinical trials, ketamine has demonstrated rapid antidepressant action in patients with treatment-resistant MDD.4,5
Recently, Murrough et al6 performed the first randomized, psychoactive controlled trial using a single IV infusion of ketamine dosed below anesthesia ranges (0.5 mg/kg), or midazolam (0.045 mg/kg), in patients with treatment-resistant depression who had been antidepressant-free for at least 4 weeks. They found that 24 hours after medication administration, the likelihood of response to ketamine was significantly higher than the response to midazolam (OR: 2.18; 95% CI: 1.21 to 4.14), with a response rate of 64% in the ketamine group and 28% in the midazolam group.6
Psychotropic side effects, such as hallucinations, are a major concern with ketamine tolerability and abuse potential. This is largely because of ketamine’s antagonism of the NMDA receptor, which is a property shared with other abused drugs such as phencyclidine (PCP) and dextromethorphan. In the Murrough et al6 study, there were no reported cases of paranoia or hallucinations, but dissociative symptoms were relatively common (17%).
Although the results in this trial appear encouraging, there are several limitations to using ketamine to treat MDD, especially in an ambulatory setting. Concerns include ketamine’s IV administration, potential for abuse, long-term efficacy, and side-effect profile—particularly psychotic symptoms and hemodynamic changes. An ideal compound would have the rapid efficacy of ketamine, but with a safer side-effect profile, easier administration, and less potential for abuse.
Riluzole also acts on the glutamatergic system, but has not shown antidepressant efficacy as consistently as ketamine. Riluzole is FDA-approved for treating amyotrophic lateral sclerosis.5 Pharmacologically, riluzole is a glutamatergic modulator that increases glutamate reuptake into glial cells, decreases glutamate release, and increases AMPA trafficking. In open-label studies riluzole has shown efficacy in reducing depressive symptoms.4,5 However, when compared with placebo as a means of sustaining treatment response after a 1-time dose of ketamine, riluzole showed was no significant improvement in time to depressive relapse.7
Acamprosate, often used for treating alcohol abuse, is another a drug with glutamatergic activity that has been studied for possible use as an antidepressant.5
A review by Lapidus et al5 has a more extensive listing of current medications and investigational compounds that modulate glutamate transmission, and are of interest for their possible antidepressant activity. Given the relatively new “glutamatergic hypothesis” of depression, it is exciting that so many current and novel glutamatergic drug therapies are being evaluated.
Future of ketamine treatment
Glutamate has been shown to play an important part in the pathophysiology of depression. The rapid antidepressant efficacy of ketamine provides evidence that future medications with glutamate-modulating activity could be useful for patients who struggle to achieve symptom relief using available antidepressants. Several limitations exist regarding ketamine use, and more work in this important therapeutic area needs to be done. This last point is important to remember when speaking with patients such as Mrs. S. Although it is understandable for her to be excited about novel treatment options such as ketamine, stress to her that treating depression with ketamine at this time is strictly investigational, and that the drug needs to be thoroughly evaluated for safety and efficacy before it can be prescribed for this indication.
CASE CONTINUED
Mrs. S realizes that ketamine may not be the best next step for her, and she agrees to explore other approaches to treat her residual depressive symptoms.
Related Resources
• Machado-Vieira R, Ibrahim L, Henter ID, et al. Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol Biochem Behav. 2012;100(4):678-687.
• Mathews DC, Henter ID, Zarate CA. Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs. 2012;72(10):1313-1333.
Drug Brand Names
Acamprosate • Campral Duloxetine • Cymbalta
Aripiprazole • Abilify Ketamine • Ketalar
Bupropion • Wellbutrin, Zyban Riluzole • Rilutek
Disclosures
The authors report no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products.
Mrs. S, age 46, has been struggling to manage depression for 7 years. She completed adequate trials of several selective serotonin reuptake inhibitors and bupropion. Currently, she is taking duloxetine, 60 mg/d, and aripiprazole, 5 mg/d.
At her most recent clinic visit, Mrs. S reports that she is doing “OK,” but that she still feels sad and disengaged most days of the week. She wants to know more about ketamine for treating depression after reading about it on the Internet and hearing it mentioned in a support group she attends. She asks if you think it would work for her, and gives you with a copy of an article about its use in patients with treatment-resistant depression. Mrs. S has no other health conditions and takes a daily vitamin D and calcium supplement.
The monoamine hypothesis of depression postulates that symptoms originate from underactivity of monoamines, such as serotonin, norepinephrine, and dopamine, in the brain. This hypothesis was formulated in the 1960s after researchers observed that monoamine oxidase inhibitors and tricyclic antidepressants relieved depressive symptoms; both were known to increase monoamine concentrations in the synaptic cleft.1
Regrettably, these medications do not adequately relieve depressive symptoms for many people. In fact, symptom remission occurs in only one-third of treated patients.2 This low remission rate reflects a lack of understanding of the pathophysiology of depression, and the need for drugs with unique mechanisms of action.
One of the newest drug targets shown to be relevant in psychiatric illness is the
glutamatergic system. Glutamate is the predominant excitatory neurotransmitter in the CNS, and it is responsible for many key functions, including synaptic plasticity, learning, memory, and locomotion.3 Normally, the glutamatergic system tightly regulates the amount of glutamate in the neuronal synapse via receptors on presynaptic and postsynaptic neurons, as well as on glial cells (Figure). When this equilibrium is disrupted in stressful situations, such as ischemia, trauma, or seizures, excess glutamate is released into the synapse. The resulting glutamatergic hyperactivity can lead to neurotoxicity and cell death when neuronal receptors are activated for an extended period.
A key component of the glutamatergic system that is responsible for removing excess glutamate from the synapse is membrane-bound transporters, which are similar to serotonin and norepinephrine transporters. These excitatory amino acid transporters (EAATs) are important because glutamate metabolism does not occur within the synapse and EAATS are responsible for removing most of the glutamate from the synapse into glial cells.3
The network of receptors within the synapse that are activated by glutamate is extensive and complex. There are at least 11 glutamate-responsive receptors: 3 are ionotropic action channels, and the remaining 8 are metabotropic G protein-coupled receptors. Previous studies have shown regional changes in glutamate receptors, as well as elevated levels of glutamate, in the brains of patients with major depressive disorder (MDD).4
Ketamine. The ionotropic receptor N-methyl-d-aspartate (NMDA) is one of the most studied glutamate receptors. Pharmacologically, ketamine is a noncompetitive NMDA receptor antagonist that also activates the amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, which is another subtype of ionotropic glutamate receptors. In open-label clinical trials, ketamine has demonstrated rapid antidepressant action in patients with treatment-resistant MDD.4,5
Recently, Murrough et al6 performed the first randomized, psychoactive controlled trial using a single IV infusion of ketamine dosed below anesthesia ranges (0.5 mg/kg), or midazolam (0.045 mg/kg), in patients with treatment-resistant depression who had been antidepressant-free for at least 4 weeks. They found that 24 hours after medication administration, the likelihood of response to ketamine was significantly higher than the response to midazolam (OR: 2.18; 95% CI: 1.21 to 4.14), with a response rate of 64% in the ketamine group and 28% in the midazolam group.6
Psychotropic side effects, such as hallucinations, are a major concern with ketamine tolerability and abuse potential. This is largely because of ketamine’s antagonism of the NMDA receptor, which is a property shared with other abused drugs such as phencyclidine (PCP) and dextromethorphan. In the Murrough et al6 study, there were no reported cases of paranoia or hallucinations, but dissociative symptoms were relatively common (17%).
Although the results in this trial appear encouraging, there are several limitations to using ketamine to treat MDD, especially in an ambulatory setting. Concerns include ketamine’s IV administration, potential for abuse, long-term efficacy, and side-effect profile—particularly psychotic symptoms and hemodynamic changes. An ideal compound would have the rapid efficacy of ketamine, but with a safer side-effect profile, easier administration, and less potential for abuse.
Riluzole also acts on the glutamatergic system, but has not shown antidepressant efficacy as consistently as ketamine. Riluzole is FDA-approved for treating amyotrophic lateral sclerosis.5 Pharmacologically, riluzole is a glutamatergic modulator that increases glutamate reuptake into glial cells, decreases glutamate release, and increases AMPA trafficking. In open-label studies riluzole has shown efficacy in reducing depressive symptoms.4,5 However, when compared with placebo as a means of sustaining treatment response after a 1-time dose of ketamine, riluzole showed was no significant improvement in time to depressive relapse.7
Acamprosate, often used for treating alcohol abuse, is another a drug with glutamatergic activity that has been studied for possible use as an antidepressant.5
A review by Lapidus et al5 has a more extensive listing of current medications and investigational compounds that modulate glutamate transmission, and are of interest for their possible antidepressant activity. Given the relatively new “glutamatergic hypothesis” of depression, it is exciting that so many current and novel glutamatergic drug therapies are being evaluated.
Future of ketamine treatment
Glutamate has been shown to play an important part in the pathophysiology of depression. The rapid antidepressant efficacy of ketamine provides evidence that future medications with glutamate-modulating activity could be useful for patients who struggle to achieve symptom relief using available antidepressants. Several limitations exist regarding ketamine use, and more work in this important therapeutic area needs to be done. This last point is important to remember when speaking with patients such as Mrs. S. Although it is understandable for her to be excited about novel treatment options such as ketamine, stress to her that treating depression with ketamine at this time is strictly investigational, and that the drug needs to be thoroughly evaluated for safety and efficacy before it can be prescribed for this indication.
CASE CONTINUED
Mrs. S realizes that ketamine may not be the best next step for her, and she agrees to explore other approaches to treat her residual depressive symptoms.
Related Resources
• Machado-Vieira R, Ibrahim L, Henter ID, et al. Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol Biochem Behav. 2012;100(4):678-687.
• Mathews DC, Henter ID, Zarate CA. Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs. 2012;72(10):1313-1333.
Drug Brand Names
Acamprosate • Campral Duloxetine • Cymbalta
Aripiprazole • Abilify Ketamine • Ketalar
Bupropion • Wellbutrin, Zyban Riluzole • Rilutek
Disclosures
The authors report no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products.
1. Niciu MJ, Ionescu DF, Richards EM, et al. Glutamate and its receptors in the pathophysiology and treatment of major depressive disorder. J Neural Transm. 2014;121(8):907-924.
2. Gaynes BN, Dusetzina SB, Ellis AR, et al. Treating depression after initial treatment failure: directly comparing switch and augmenting strategies in STAR*D. J Clin Psychopharmacol. 2012;32(1):114-119.
3. Curry SC, Mills KC, Ruha A, et al. Neurotransmitters and neuromodulators. In: Nelson LS, Lewin NA, Howland MA, et al, eds. Goldfrank’s toxicologic emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:189-220.
4. Zarate C Jr, Machado-Vieira R, Henter I, et al. Glutamatergic modulators: the future of treating mood disorders? Harv Rev Psychiatry. 2010;18(5):293-303.
5. Lapidus KA, Soleimani L, Murrough JW. Novel glutamatergic drugs for the treatment of mood disorders. Neuropsychiatr Dis Treat. 2013;9:1101-1112.
6. Murrough JW, Iosifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
7. Ibrahim L, Diazgranados N, Franco-Chaves J, et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology. 2012;37(6):1526-1533.
1. Niciu MJ, Ionescu DF, Richards EM, et al. Glutamate and its receptors in the pathophysiology and treatment of major depressive disorder. J Neural Transm. 2014;121(8):907-924.
2. Gaynes BN, Dusetzina SB, Ellis AR, et al. Treating depression after initial treatment failure: directly comparing switch and augmenting strategies in STAR*D. J Clin Psychopharmacol. 2012;32(1):114-119.
3. Curry SC, Mills KC, Ruha A, et al. Neurotransmitters and neuromodulators. In: Nelson LS, Lewin NA, Howland MA, et al, eds. Goldfrank’s toxicologic emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:189-220.
4. Zarate C Jr, Machado-Vieira R, Henter I, et al. Glutamatergic modulators: the future of treating mood disorders? Harv Rev Psychiatry. 2010;18(5):293-303.
5. Lapidus KA, Soleimani L, Murrough JW. Novel glutamatergic drugs for the treatment of mood disorders. Neuropsychiatr Dis Treat. 2013;9:1101-1112.
6. Murrough JW, Iosifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
7. Ibrahim L, Diazgranados N, Franco-Chaves J, et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology. 2012;37(6):1526-1533.