User login
In reply: Myasthenia gravis
In Reply: We appreciate Dr. Keiter’s comments. We agree that myasthenia gravis, like most medical disorders, rests on clinical diagnosis. We have patients we treat for myasthenia gravis in the absence of the typical serological confirmation. A very few of these patients with restricted oculobulbar symptoms may also have normal single-fiber EMG studies. In this situation, the decision to treat an individual for myasthenia gravis must rest on the physician’s clinical judgment, but also on the patient’s understanding that the condition does not have the diagnostic support often seen. The decision to treat with medications that have potential severe side effects requires the patient’s understanding of the context in which the diagnosis is being made and the specific treatment is being suggested.
In Reply: We appreciate Dr. Keiter’s comments. We agree that myasthenia gravis, like most medical disorders, rests on clinical diagnosis. We have patients we treat for myasthenia gravis in the absence of the typical serological confirmation. A very few of these patients with restricted oculobulbar symptoms may also have normal single-fiber EMG studies. In this situation, the decision to treat an individual for myasthenia gravis must rest on the physician’s clinical judgment, but also on the patient’s understanding that the condition does not have the diagnostic support often seen. The decision to treat with medications that have potential severe side effects requires the patient’s understanding of the context in which the diagnosis is being made and the specific treatment is being suggested.
In Reply: We appreciate Dr. Keiter’s comments. We agree that myasthenia gravis, like most medical disorders, rests on clinical diagnosis. We have patients we treat for myasthenia gravis in the absence of the typical serological confirmation. A very few of these patients with restricted oculobulbar symptoms may also have normal single-fiber EMG studies. In this situation, the decision to treat an individual for myasthenia gravis must rest on the physician’s clinical judgment, but also on the patient’s understanding that the condition does not have the diagnostic support often seen. The decision to treat with medications that have potential severe side effects requires the patient’s understanding of the context in which the diagnosis is being made and the specific treatment is being suggested.
Myasthenia gravis: Newer therapies offer sustained improvement
Current therapies for myasthenia gravis can help most patients achieve sustained improvement. The overall prognosis has dramatically improved over the last 4 decades: the mortality rate used to be 75%; now it is 4.5%.1
Myasthenia gravis is the most common disorder of neuromuscular junction transmission and is also one of the best characterized autoimmune diseases. However, its symptoms—primarily weakness—vary from patient to patient, and in the same patient, by time of day and over longer time periods. The variation in symptoms can be very confusing to undiagnosed patients and puzzling to unsuspecting physicians. Such diagnostic uncertainty can give the patient additional frustration and emotional stress, which in turn exacerbate his or her condition.
In this review, we will give an overview of the pathogenesis, clinical manifestations, diagnosis, and treatment of myasthenia gravis.
TWO PEAKS IN INCIDENCE BY AGE

The annual incidence of myasthenia gravis is approximately 10 to 20 new cases per million, with a prevalence of about 150 to 200 per million.2
The age of onset has a bimodal distribution, with an early incidence peak in the second to third decade with a female predominance and a late peak in the 6th to the 8th decade with a male predominance.2
Myasthenia gravis is commonly associated with several other autoimmune disorders, including hypothyroidism, hyperthyroidism, systemic lupus erythematosus, rheumatoid arthritis, vitiligo, diabetes, and, more recently recognized, neuromyelitis optica.3
ANTIBODIES AGAINST AChR AND MuSK
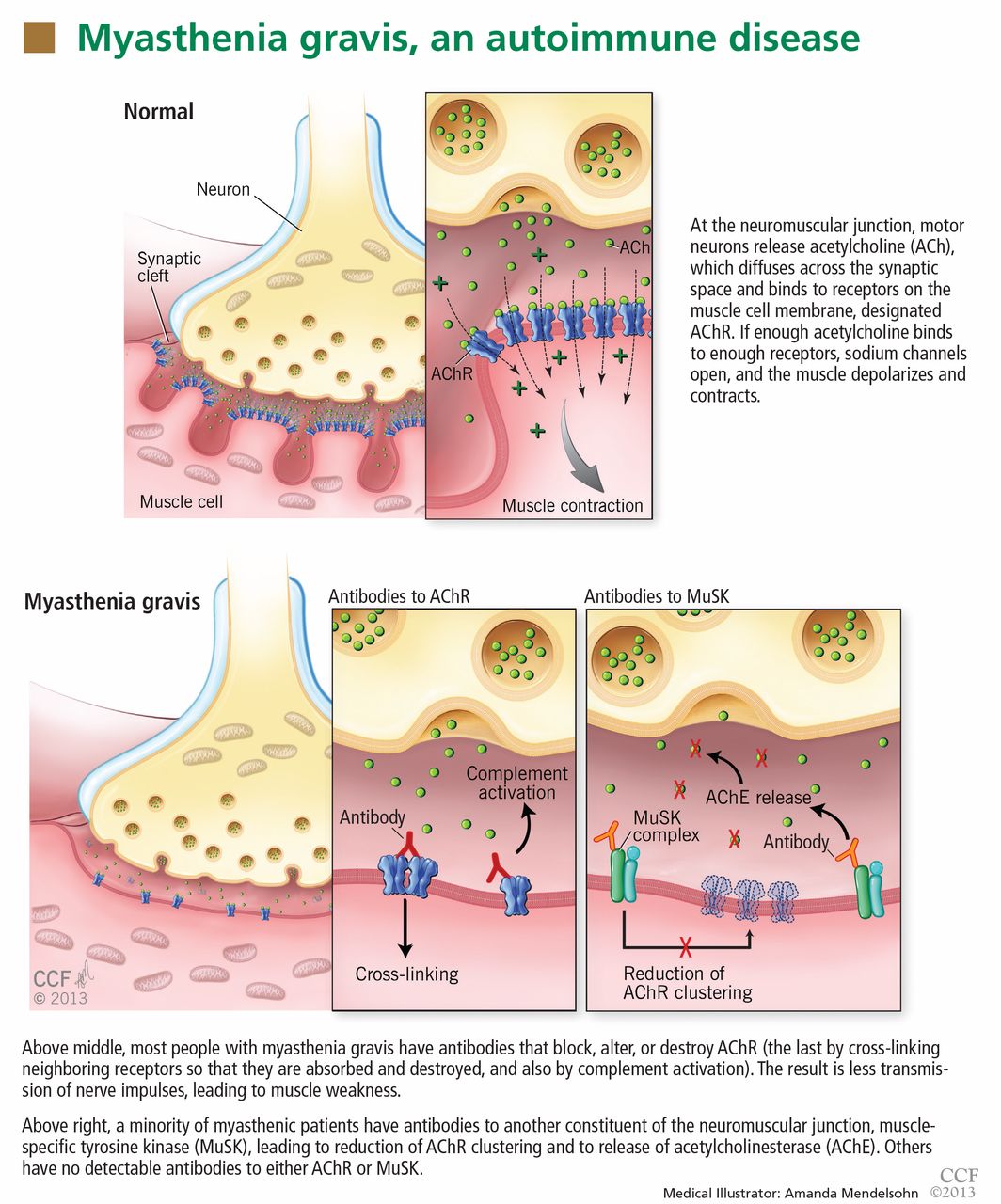
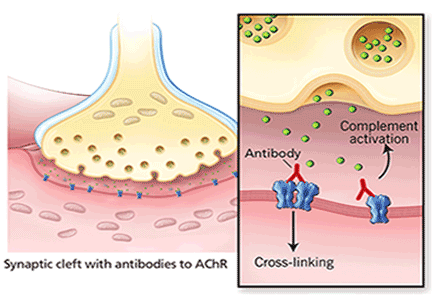
In most cases of myasthenia gravis the patient has autoimmune antibodies against constituents of the neuromuscular junction, specifically acetylcholine receptor (AChR) and muscle-specific tyrosine kinase (MuSK) (Figure 1).
AChR antibody-positive myasthenia gravis
When antibodies bind to AChR on the postsynaptic membrane, they cross-link neighboring AChR units, which are absorbed into the muscle fiber and are broken up.4 In addition, the complement system is activated to mediate further damage on the postsynaptic membrane.
AChR antibodies may come from germinal centers of the thymus, where clustered myoid cells express AChR on the plasma membrane surface.5 About 60% of AChR antibody-positive myasthenia gravis patients have an enlarged thymus, and 10% have a thymoma—a tumor of the epithelial cells of this organ. Conversely, about 15% of patients with a thymoma have clinical myasthenia gravis, and an additional 20% possess antibodies against AChR in the serum without myasthenic symptoms.5
MuSK antibody-positive myasthenia gravis
Like AChR, MuSK is a transmembrane component of the postsynaptic neuromuscular junction. During formation of the neuromuscular junction, MuSK is activated through the binding of agrin (a nerve-derived proteoglycan) to lipoprotein-related protein 4 (LRP4), after which complicated intracellular signaling promotes the assembly and stabilization of AChR.6
Unlike AChR antibodies, antibodies against MuSK do not activate the complement system, and complement fixation is not essential for clinical myasthenic symptoms to appear.7 Also, myasthenia gravis with MuSK antibodies is rarely associated with thymoma.8
The precise mechanism by which MuSK antibody impairs transmission at the neuromuscular junction has been a mystery until recently. Animal models, including MuSK-mutant mice and mice injected with MuSK protein or with purified immunoglobulin G from patients with this disease, have revealed a significant reduction of AChR clusters and destruction of neuromuscular junction structures.7,9–12
In addition, MuSK antibodies produce pre-synaptic dysfunction, manifesting as a reduction of acetylcholine content. This information is based on studies in mice and on in vitro electrophysiologic analyses of neuromuscular junctions from a patient with this disease.7,9–13
Finally, MuSK antibodies may indirectly affect the recycling of acetylcholine. After post-synaptic activation, acetylcholine is normally hydrolized by acetylcholinesterase, which is located in the synaptic cleft but anchored to MuSK on the postsynaptic membrane. MuSK antibodies block the binding of MuSK to acetylcholinesterase, possibly leading to less accumulation of acetylcholinesterase.14 This process may explain why patients with MuSK antibody-positive myasthenia gravis tend to respond poorly to acetylcholinesterase inhibitors (more about this below).
Seronegative myasthenia gravis
In a series of 562 consecutive patients with generalized weakness due to myasthenia gravis, 92% were positive for AChR antibody, 3% were positive for MuSK antibody, and 5% were seronegative (possessing neither antibody).15 In contrast, about 50% of patients with purely ocular myasthenia gravis (ie, with isolated weakness of the levator palpebrae superioris, orbicularis oculi, or oculomotor muscles) are seropositive for AChR antibody. Only a few ocular MuSK antibody-positive cases have been described, leaving the rest seronegative. Rarely, both antibodies can be detected in the same patient.16
In patients who are negative for AChR antibodies at the time of disease onset, sero-conversion may occur later during the course. Repeating serologic testing 6 to 12 months later may detect AChR antibodies in approximately 15% of patients who were initially seronegative.15,17
The clinical presentation, electrophysiologic findings, thymic pathologic findings, and treatment responses are similar in AChR antibody-positive and seronegative myasthenia gravis.17 Muscle biopsy study in seronegative cases demonstrates a loss of AChR as well.18
Based on these observations, it has been proposed that seronegative patients may have low-affinity antibodies that can bind to tightly clustered AChRs on the postsynaptic membrane but escape detection by routine radioimmunoassays in a solution phase. With a sensitive cell-based immunofluorescence assay, low-affinity antibodies to clustered AChRs were detected in 66% of patients with generalized myasthenia gravis and in 50% of those with ocular myasthenia gravis who were seronegative on standard assays.19,20 These low-affinity AChR antibodies can also activate complement in vitro, increasing the likelihood that they are pathogenic. However, assays to detect low-affinity AChR antibodies are not yet commercially available.
Within the past year, three research groups independently reported detecting antibodies to LRP4 in 2% to 50% of seronegative myasthenia gravis patients. This wide variation in the prevalence of LRP4 antibodies could be related to patient ethnicity and methods of detection.21–23 LRP4 is a receptor for agrin and is required for agrin-induced MuSK activation and AChR clustering. LRP antibodies can activate complement; therefore, it is plausible that LRP4 antibody binding leads to AChR loss on the postsynaptic membrane. However, additional study is needed to determine if LRP4 antibodies are truly pathogenic in myasthenia gravis.
A DISORDER OF FATIGABLE WEAKNESS

Myasthenia gravis is a disorder of fatigable weakness producing fluctuating symptoms. Symptoms related to the involvement of specific muscle groups are listed in Table 1. Muscle weakness is often worse later in the day or after exercise.
Ocular myasthenia gravis accounts for about 15% of all cases. Of patients initially presenting with ocular symptoms only, twothirds will ultimately develop generalized symptoms, most within the first 2 years.24 No factor has been identified that predicts conversion from an ocular to a generalized form.
Several clinical phenotypes of MuSK antibody-positive myasthenia gravis have been described. An oculobulbar form presents with diplopia, ptosis, dysarthria, and profound atrophy of the muscles of the tongue and face. A restricted myopathic form presents with prominent neck, shoulder, and respiratory weakness without ocular involvement. A third form is a combination of ocular and proximal limb weakness, indistinguishable from AChR antibody-positive disease.25
MuSK antibody-positive patients do not respond as well to acetylcholinesterase inhibitors as AChR antibody-positive patients do. In one study, nearly 70% of MuSK antibody-positive patients demonstrated no response, poor tolerance, or cholinergic hypersensitivity to these agents.25 Fortunately, most MuSK antibody-positive patients have a favorable response to immunosuppressive therapy—sometimes a dramatic improvement after plasmapheresis.8
DIAGNOSIS OF MYASTHENIA GRAVIS
The common differential diagnoses for myasthenia gravis are listed in Table 2.
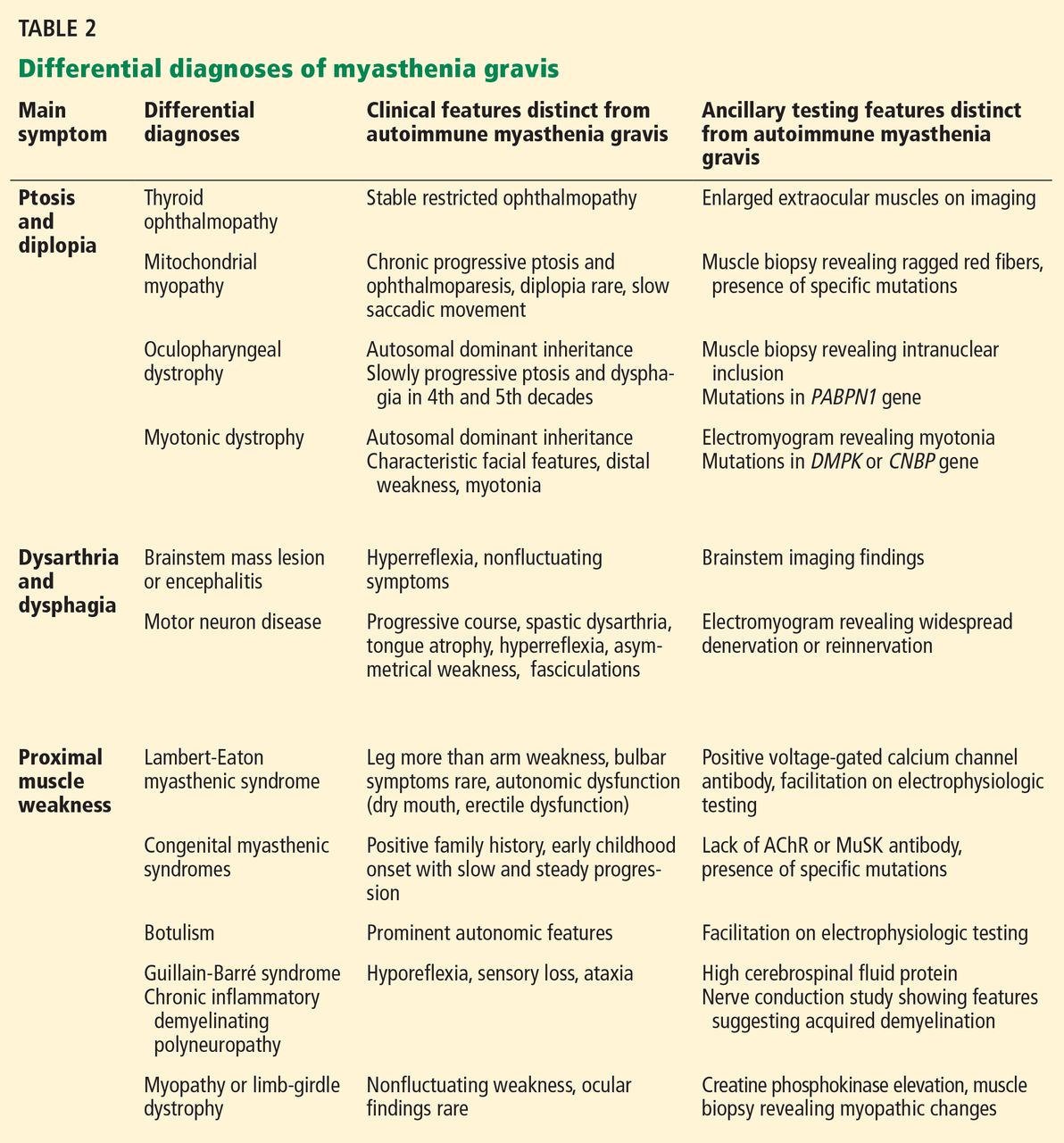
The essential feature of myasthenia gravis is fluctuating muscle weakness, often with fatigue. Many patients complain of weakness of specific muscle groups after their repeated usage. Pain is generally a less conspicuous symptom, and generalized fatigue without objective weakness is inconsistent with myasthenia gravis.
Signs of muscle weakness may include droopy eyelids, diplopia, inability to hold the head straight, difficulty swallowing or chewing, speech disturbances, difficulty breathing, and difficulty raising the arms or rising from the sitting position. A historical pattern of ptosis alternating from one eye to the other is fairly characteristic of myasthenia gravis.
The weakness of orbicularis oculi is easily identified on examination by prying open the eyes during forced eye closure. Limb weakness is usually more significant in the arms than in the legs. An often-neglected feature of myasthenia gravis is finger extensor weakness with a relative sparing of other distal hand muscles.2
The ice-pack test is performed by placing a small bag of ice over the ptotic eye for 2 to 5 minutes and assessing the degree of ptosis for any noticeable improvement. This test is not very helpful for assessing ocular motor weakness.
The edrophonium (Tensilon) test can be used for patients with ptosis or ophthalmoparesis. Edrophonium, a short-acting acetylcholinesterase inhibitor, is given intravenously while the patient is observed for objective improvement. The patient’s cardiovascular status should be monitored for arrhythmias and hypotension. Atropine should be immediately available in case severe bradycardia develops.
The ice-pack test and the edrophonium test can give false-negative and false-positive results, and the diagnosis of myasthenia gravis must be verified by other diagnostic tests.
Testing for antibodies
Testing for circulating AChR antibodies, MuSK antibodies, or both is the first step in the laboratory confirmation of myasthenia gravis.
There are three AChR antibody subtypes: binding, blocking, and modulating. Binding antibodies are present in 80% to 90% of patients with generalized myasthenia gravis and 50% of those with ocular myasthenia gravis. Testing for blocking and modulating AChR antibodies increases the sensitivity by less than 5% when added to testing for binding antibodies.
AChR antibody titers correlate poorly with disease severity between patients. However, in individual patients, antibody titers tend to go down in parallel with clinical improvement.
MuSK antibody is detected in nearly half of myasthenia gravis patients with generalized weakness who are negative for AChR antibody.
Electrophysiologic tests
Electrophysiologic tests can usually confirm the diagnosis of seronegative myasthenia gravis. They are also helpful in seropositive patients who have unusual clinical features or a poor response to treatment.
Repetitive nerve stimulation studies use a slow rate (2–5 Hz) of repetitive electrical stimulation. The study is positive if the motor response declines by more than 10%. However, a decremental response is not specific for myasthenia gravis, as it may be seen in other neuromuscular disorders such as motor neuron disease or Lambert-Eaton myasthenic syndrome.
This test is technically easier to do in distal muscles than in proximal muscles, but less sensitive. Therefore, proximal muscles such as the trapezius or facial muscles are usually also sampled to maximize the yield. To further maximize the sensitivity, muscles being tested should be warm, and acetylcholinesterase inhibitors should be withheld for 12 hours before.
Repetitive nerve stimulation studies in distal muscles are positive in approximately 75% of patients with generalized myasthenia gravis and in 30% with ocular myasthenia gravis.26
Single-fiber electromyography is more technically demanding than repetitive nerve stimulation and is less widely available. It is usually performed with a special needle electrode that can simultaneously identify action potentials arising from individual muscle fibers innervated by the same axon.
Variability in time of the second action potential relative to the first is called “jitter.” Abnormal jitter is seen in more than 95% of patients with generalized myasthenia gravis and in 85% to 90% of those with ocular myasthenia gravis.26,27 However, abnormal jitter can also be seen in other neuromuscular diseases such as motor neuron disease or in neuromuscular junctional disorders such as Lambert-Eaton myasthenic syndrome.
Imaging studies
Chest computed tomography or magnetic resonance imaging with contrast should be performed in all myasthenia gravis patients to look for a thymoma.
TREATMENT OF MYASTHENIA GRAVIS
Acetylcholinesterase inhibitors
As a reasonable first therapy in mild cases of myasthenia gravis, acetylcholinesterase inhibitors slow down the degradation of acetylcholine and prolong its effect in the neuromuscular junction, but they are not disease-modifying and their benefits are mild.
Pyridostigmine is the usual choice of acetylcholinesterase inhibitor. Its onset of action is rapid (15 to 30 minutes) and its action lasts for 3 to 4 hours. For most patients, the effective dosage range is 60 mg to 90 mg every 4 to 6 hours. A long-acting form is also available and can be given as a single nighttime dose.
Immunomodulating therapy
Patients who have moderate to severe symptoms require some form of immunomodulating therapy.
Plasmapheresis or intravenous immune globulin is reserved for patients with severe or rapidly worsening disease because their beneficial effects can be seen within the first week of treatment.
Longer-acting immunotherapies (corticosteroids, azathioprine, mycophenolate mofetil and others) have a slower onset of responses but provide sustained benefits. Which drug to use depends on factors such as comorbidity, side effects, and cost.
Drugs to avoid
A number of medications can exacerbate weakness in myasthenia gravis and should be avoided or used with caution. The list is long, but ones that deserve the most attention are penicillamine, interferons, procainamide, quinidine, and antibiotics, including quinolones and aminoglycosides. A more comprehensive list of medications that may exacerbate myasthenia gravis symptoms can be found in a review by Keesey.2
RAPID INDUCTION IMMUNOTHERAPIES : PLASMAPHERESIS, IMMUNE GLOBULIN
Both plasmapheresis and intravenous immune globulin act quickly over days, but in most patients their effects last only a few weeks. Both are used as rescue therapies for myasthenic crises, bridging therapy to slow-acting immunotherapeutic agents, or maintenance treatment for poorly controlled cases.
Several retrospective studies have confirmed the efficacy of plasmapheresis in more than 80% of patients with generalized symptoms.28,29
In a randomized trial in patients with generalized therapies, intravenous immune globulin improved muscle strength in the group of patients with severe symptoms.30 The effective dosage of intravenous immune globulin varies from 1 to 2 g/kg without observed difference between doses.31 Trials comparing the efficacy of intravenous immune globulin and plasmapheresis in acute and severe myasthenia gravis did not reveal a difference in efficacy.32,33 Intravenous immune globulin at a minimal dose of 0.4 g/kg every 3 months has been successfully used as a long-term maintenance monotherapy, and such a role could be expanded to more patients with further studies.34
The choice between plasmapheresis and intravenous immune globulin is often based on the ability of a patient to tolerate each treatment and on the availability of the plasmapheresis procedure. Intravenous immune globulin is easier to administer, is associated with fewer adverse events related to vascular access, and is therefore more appropriate than plasmapheresis in some centers.
CHRONIC MAINTENANCE IMMUNOMODULATING TREATMENT
Corticosteroids
Prednisone, the most commonly used agent, leads to remission or marked improvement in 70% to 80% of patients with ocular or generalized myasthenia gravis.35 It may also reduce the progression of ocular myasthenia gravis to the generalized form.36
The effective dose of prednisone depends on the severity and distribution of symptoms. Some patients may need up to 1.0 mg/kg/day (usually 50 to 80 mg per day). In patients with mild to moderate symptoms, a lower maximal dosage such as 20 to 40 mg per day can be sufficient.
Within 1 to 2 weeks after starting high-dose prednisone, up to 50% of patients may develop a transient deterioration, including possible precipitation of a myasthenic crisis.37 For this reason, high-dose prednisone is commonly started only in hospitalized patients who are also receiving plasmapheresis or intravenous immune globulin. Otherwise, an outpatient dose-escalation protocol can be used to achieve a target dose over several weeks.
Prednisone tapering can begin after the patient has been on the maximal dose for 1 to 2 months and significant improvement is evident. A monthly tapering of 5 to 10 mg is preferred, then more slowly after the daily dose reaches 30 mg. The usual maintenance dose averages about 5 mg daily.
Common side effects of prednisone include weight gain, cushingoid features, easy bruising, cataracts, glaucoma, hypertension, diabetes, dyslipidemia, and osteoporosis. Patients are advised to take supplemental calcium (1,500 mg per day) and vitamin D (400 to 800 IU per day). For those most at risk of osteoporosis, treatment with a bisphosphonate should be considered.
Other immunotherapeutic agents are often needed, either to replace the corticosteroid or to permit use of lower doses of it. Because of their delayed onset of action, starting such corticosteroid-sparing agents early in the course is often necessary. These agents are often initially combined with high-dose prednisone, with an eventual goal of weaning off prednisone entirely. This strategy offers the advantage of relatively rapid induction while avoiding the long-term adverse effects of corticosteroid treatment.
Azathioprine
Azathioprine doesn’t begin to show a beneficial effect in myasthenia gravis for 6 to 12 months, and it often reaches its maximal efficacy only after 1 to 2 years of treatment.38
In a study of 78 myasthenia gravis patients, 91% improved when treated with azathioprine alone or together with prednisone.39 In another study using azathioprine and prednisolone for generalized myasthenia gravis, nearly two-thirds of patients came off prednisolone while maintaining remission for 3 years.38
A typical maintenance dose is 2 to 3 mg/kg/day. Common side effects are nausea, vomiting, and malaise. Less frequent side effects include hematologic abnormalities, abnormal liver function, and pancreatitis. Monthly monitoring of complete blood cell counts and liver function tests is warranted for the first 6 months, then less often.
One in 300 people in the general population is homozygous for a mutant allele in the thiopurine methyltransferase (TPMT) gene. Patients with this genotype should not receive azathioprine because of the risk of life-threatening bone marrow suppression.40 A slightly increased risk of various forms of lymphoma has been documented.41
Mycophenolate mofetil
A well-tolerated medication with few side effects, mycophenolate mofetil is being used more in myasthenia gravis. The results of two recent randomized trials suggested that it is not effective in improving myasthenia gravis symptoms or sparing prednisone dosage when used for 90 days or 36 weeks.42,43 However, extensive clinical experience supports its longterm efficacy in myasthenia gravis.
In a retrospective study of 85 patients with generalized myasthenia gravis, mycophenolate at doses of 1 to 3 g daily improved symptoms in 73% and produced remission in 50%. Steroid dosage was reduced in 71% of patients.44
Another retrospective study, with 102 patients, verified a slow development of clinical benefit after months of mycophenolate therapy alone or in combination with prednisone. Approximately 50% of patients achieved a minimal manifestation status after 6 to 12 months of mycophenolate treatment. Eventually, at 24 months of treatment, 80% of patients had a desirable outcome of minimal clinical manifestation or better, 55% of patients were able to come off prednisone entirely, and 75% were taking less than 7.5 mg of prednisone per day.45
Common side effects of mycophenolate include nausea, diarrhea, and infections such as urinary tract infections and herpes reactivation. The complete blood cell count needs to be monitored frequently during the first 6 months of therapy. Leukopenia can occur but rarely necessitates stopping mycophenolate. Long-term safety data are lacking, but so far there has been no clearly increased risk of malignancy.
Mycophenolate exposure in pregnancy results in a high incidence of major fetal malformations. Therefore, its use in pregnant patients is discouraged, and women of child-bearing age should use effective contraception.46
Cyclosporine
A randomized trial in a small number of patients suggested that cyclosporine is fairly effective as monotherapy.47 Its onset of action in myasthenia gravis is faster than that of other corticosteroid-sparing agents, and clinical benefit can often be observed as early as 1 to 2 months. A dose of 5 mg/kg/day and a maintenance serum level of 100 to 150 ng/mL are generally recommended. However, renal, hepatic, and hematologic toxicities and interactions with other medications make cyclosporine a less attractive choice.
Methotrexate
A randomized trial evaluated the utility of methotrexate as a steroid-sparing agent compared with azathioprine.48 At 24 months, its steroid-sparing effect was similar to that of azathioprine, and the prednisone dosage had been reduced in more than 50% of patients.
Another phase II trial studying the efficacy of methotrexate in myasthenia gravis is under way.49
Rituximab
Rituximab is a monoclonal antibody against B-cell membrane marker CD20. A growing number of case series support its efficacy in patients with severe generalized myasthenia gravis refractory to multiple immunosuppressants.16,50 It seems particularly effective for MuSK antibody-positive disease, reducing MuSK antibody titers and having a treatment effect that lasts for years.
The standard dosage is 375 mg/m2 per week for 4 consecutive weeks. Peripheral B cells tend to be depleted within 2 weeks after the first infusion, while T-cell populations remain unchanged.50
A minimal infusion reaction such as flushing and chills can be seen with the first infusion. Patients may be more susceptible to certain infections such as reactivation of herpes zoster, but overall rituximab is well tolerated. Rare cases of progressive multifocal leukoencephalopathy have been reported in patients taking it, but none have occurred so far in myasthenia gravis treatment.
Cyclophosphamide
Cyclophosphamide is an alkylating agent that reduces proliferation of both B and T cells. It can be effective in myasthenia gravis, but potentially serious side effects limit its use. It should be reserved for the small percentage of cases that are refractory to other immunotherapies.
Thymectomy
Surgical treatment should be considered for patients with thymoma. If the tumor cannot be surgically resected, chemoradiotherapy can be considered for relief of myasthenic symptoms and for prevention of local invasion.
Thymomas recur in a minority of patients many years after the initial resection, sometimes without myasthenia gravis symptoms. A recurrence of symptoms does not necessarily indicate a recurrence of thymoma. The lack of correlation between myasthenia gravis symptoms and thymoma recurrence highlights the importance of radiologic follow-up in these patients.
For patients without thymoma, many experts believe that thymectomy is beneficial in patients under age 60 who have generalized myasthenia gravis. The likelihood of medication-free remission is about twice as high, and the likelihood of becoming asymptomatic is about one and a half times higher after thymectomy.51 However, it takes up to several years for the benefits of thymectomy to manifest, and thymectomy does not guarantee protection from developing AChR antibody-positive myasthenia gravis in the future.
The optimal timing of thymectomy is not well established; however, the procedure is usually recommended within the first 3 years of diagnosis.52 The response rates from thymectomy are similar for AChR antibody-positive and seronegative patients. In general, thymectomy for MuSK antibody-positive patients has not been effective, and its role in ocular myasthenia gravis is unclear.2,53
- Alshekhlee A, Miles JD, Katirji B, Preston DC, Kaminski HJ. Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology 2009; 72:1548–1554.
- Keesey JC. Clinical evaluation and management of myasthenia gravis. Muscle Nerve 2004; 29:484–505.
- Leite MI, Coutinho E, Lana-Peixoto M, et al. Myasthenia gravis and neuromyelitis optica spectrum disorder: a multicenter study of 16 patients. Neurology 2012; 78:1601–1607.
- Drachman DB, Angus CW, Adams RN, Michelson JD, Hoffman GJ. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med 1978; 298:1116–1122.
- Fujii Y. The thymus, thymoma and myasthenia gravis. Surg Today 2013; 43:461–466.
- Evoli A, Lindstrom J. Myasthenia gravis with antibodies to MuSK: another step toward solving mystery? Neurology 2011; 77:1783–1784.
- Mori S, Kubo S, Akiyoshi T, et al. Antibodies against muscle-specific kinase impair both presynaptic and postsynaptic functions in a murine model of myasthenia gravis. Am J Pathol 2012; 180:798–810.
- Guptill JT, Sanders DB, Evoli A. Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve 2011; 44:36–40.
- Chevessier F, Girard E, Molgó J, et al. A mouse model for congenital myasthenic syndrome due to MuSK mutations reveals defects in structure and function of neuromuscular junctions. Hum Mol Genet 2008; 17:3577–3595.
- Richman DP, Nishi K, Morell SW, et al. Acute severe animal model of anti-muscle-specific kinase myasthenia: combined postsynaptic and presynaptic changes. Arch Neurol 2012; 69:453–460.
- Klooster R, Plomp JJ, Huijbers MG, et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain 2012; 135:1081–1101.
- Viegas S, Jacobson L, Waters P, et al. Passive and active immunization models of MuSK-Ab positive myasthenia: electrophysiological evidence for pre and postsynaptic defects. Exp Neurol 2012; 234:506–512.
- Niks EH, Kuks JB, Wokke JH, et al. Pre- and postsynaptic neuromuscular junction abnormalities in musk myasthenia. Muscle Nerve 2010; 42:283–288.
- Kawakami Y, Ito M, Hirayama M, et al. Anti-MuSK autoantibodies block binding of collagen Q to MuSK. Neurology 2011; 77:1819–1826.
- Chan KH, Lachance DH, Harper CM, Lennon VA. Frequency of seronegativity in adult-acquired generalized myasthenia gravis. Muscle Nerve 2007; 36:651–658.
- Collongues N, Casez O, Lacour A, et al. Rituximab in refractory and non-refractory myasthenia: a retrospective multicenter study. Muscle Nerve 2012; 46:687–691.
- Sanders DB, Andrews PI, Howard JF, Massey JM. Seronegative myasthenia gravis. Neurology 1997; 48(suppl 5):40S–45S.
- Shiraishi H, Motomura M, Yoshimura T, et al. Acetylcholine receptors loss and postsynaptic damage in MuSK antibody-positive myasthenia gravis. Ann Neurol 2005; 57:289–293.
- Leite MI, Jacob S, Viegas S, et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain 2008; 131:1940–1952.
- Jacob S, Viegas S, Leite MI, et al. Presence and pathogenic relevance of antibodies to clustered acetylcholine receptor in ocular and generalized myasthenia gravis. Arch Neurol 2012; 69:994–1001.
- Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol 2011; 69:418–422.
- Pevzner A, Schoser B, Peters K, et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol 2012; 259:427–435.
- Zhang B, Tzartos JS, Belimezi M, et al. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol 2012; 69:445–451.
- Kupersmith MJ, Latkany R, Homel P. Development of generalized disease at 2 years in patients with ocular myasthenia gravis. Arch Neurol 2003; 60:243–248.
- Pasnoor M, Wolfe GI, Nations S, et al. Clinical findings in MuSK-antibody positive myasthenia gravis: a US experience. Muscle Nerve 2010; 41:370–374.
- Oh SJ, Kim DE, Kuruoglu R, Bradley RJ, Dwyer D. Diagnostic sensitivity of the laboratory tests in myasthenia gravis. Muscle Nerve 1992; 15:720–724.
- Sanders DB, Stålberg EV. AAEM minimonograph #25: single-fiber electromyography. Muscle Nerve 1996; 19:1069–1083.
- Lazo-Langner A, Espinosa-Poblano I, Tirado-Cárdenas N, et al. Therapeutic plasma exchange in Mexico: experience from a single institution. Am J Hematol 2002; 70:16–21.
- Carandina-Maffeis R, Nucci A, Marques JF, et al. Plasmapheresis in the treatment of myasthenia gravis: retrospective study of 26 patients. Arq Neuropsiquiatr 2004; 62:391–395.
- Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology 2007; 68:837–841.
- Gajdos P, Tranchant C, Clair B, et al; Myasthenia Gravis Clinical Study Group. Treatment of myasthenia gravis exacerbation with intravenous immunoglobulin: a randomized double-blind clinical trial. Arch Neurol 2005; 62:1689–1693.
- Rønager J, Ravnborg M, Hermansen I, Vorstrup S. Immunoglobulin treatment versus plasma exchange in patients with chronic moderate to severe myasthenia gravis. Artif Organs 2001; 25:967–973.
- Barth D, Nabavi Nouri M, Ng E, Nwe P, Bril V. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology 2011; 76:2017–2023.
- Wegner B, Ahmed I. Intravenous immunoglobulin monotherapy in long-term treatment of myasthenia gravis. Clin Neurol Neurosurg 2002; 105:3–8.
- Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol 1984; 15:291–298.
- Monsul NT, Patwa HS, Knorr AM, Lesser RL, Goldstein JM. The effect of prednisone on the progression from ocular to generalized myasthenia gravis. J Neurol Sci 2004; 217:131–133.
- Miller RG, Milner-Brown HS, Mirka A. Prednisone-induced worsening of neuromuscular function in myasthenia gravis. Neurology 1986; 36:729–732.
- Palace J, Newsom-Davis J, Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Myasthenia Gravis Study Group. Neurology 1998; 50:1778–1783.
- Mertens HG, Hertel G, Reuther P, Ricker K. Effect of immunosuppressive drugs (azathioprine). Ann N Y Acad Sci 1981; 377:691–699.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Finelli PF. Primary CNS lymphoma in myasthenic on long-term azathioprine. J Neurooncol 2005; 74:91–92.
- Sanders DB, Hart IK, Mantegazza R, et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology 2008; 71:400–406.
- Muscle Study Group. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology 2008; 71:394–399.
- Meriggioli MN, Ciafaloni E, Al-Hayk KA, et al. Mycophenolate mofetil for myasthenia gravis: an analysis of efficacy, safety, and tolerability. Neurology 2003; 61:1438–1440.
- Hehir MK, Burns TM, Alpers J, Conaway MR, Sawa M, Sanders DB. Mycophenolate mofetil in AChR-antibody-positive myasthenia gravis: outcomes in 102 patients. Muscle Nerve 2010; 41:593–598.
- Merlob P, Stahl B, Klinger G. Tetrada of the possible mycophenolate mofetil embryopathy: a review. Reprod Toxicol 2009; 28:105–108.
- Tindall RS, Rollins JA, Phillips JT, Greenlee RG, Wells L, Belendiuk G. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med 1987; 316:719–724.
- Heckmann JM, Rawoot A, Bateman K, Renison R, Badri M. A single-blinded trial of methotrexate versus azathioprine as steroid-sparing agents in generalized myasthenia gravis. BMC Neurol 2011; 11:97.
- Pasnoor M, He J, Herbelin L, Dimachkie M, Barohn RJ; Muscle Study Group. Phase II trial of methotrexate in myasthenia gravis. Ann N Y Acad Sci 2012; 1275:23–28.
- Díaz-Manera J, Martínez-Hernández E, Querol L, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology 2012; 78:189–193.
- Gronseth GS, Barohn RJ. Practice parameter: thymectomy for autoimmune myasthenia gravis (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2000; 55:7–15.
- Kumar V, Kaminski HJ. Treatment of myasthenia gravis. Curr Neurol Neurosci Rep 2011; 11:89–96.
- Pompeo E, Tacconi F, Massa R, Mineo D, Nahmias S, Mineo TC. Long-term outcome of thoracoscopic extended thymectomy for nonthymomatous myasthenia gravis. Eur J Cardiothorac Surg 2009; 36:164–169.
Current therapies for myasthenia gravis can help most patients achieve sustained improvement. The overall prognosis has dramatically improved over the last 4 decades: the mortality rate used to be 75%; now it is 4.5%.1
Myasthenia gravis is the most common disorder of neuromuscular junction transmission and is also one of the best characterized autoimmune diseases. However, its symptoms—primarily weakness—vary from patient to patient, and in the same patient, by time of day and over longer time periods. The variation in symptoms can be very confusing to undiagnosed patients and puzzling to unsuspecting physicians. Such diagnostic uncertainty can give the patient additional frustration and emotional stress, which in turn exacerbate his or her condition.
In this review, we will give an overview of the pathogenesis, clinical manifestations, diagnosis, and treatment of myasthenia gravis.
TWO PEAKS IN INCIDENCE BY AGE
The annual incidence of myasthenia gravis is approximately 10 to 20 new cases per million, with a prevalence of about 150 to 200 per million.2
The age of onset has a bimodal distribution, with an early incidence peak in the second to third decade with a female predominance and a late peak in the 6th to the 8th decade with a male predominance.2
Myasthenia gravis is commonly associated with several other autoimmune disorders, including hypothyroidism, hyperthyroidism, systemic lupus erythematosus, rheumatoid arthritis, vitiligo, diabetes, and, more recently recognized, neuromyelitis optica.3
ANTIBODIES AGAINST AChR AND MuSK
In most cases of myasthenia gravis the patient has autoimmune antibodies against constituents of the neuromuscular junction, specifically acetylcholine receptor (AChR) and muscle-specific tyrosine kinase (MuSK) (Figure 1).
AChR antibody-positive myasthenia gravis
When antibodies bind to AChR on the postsynaptic membrane, they cross-link neighboring AChR units, which are absorbed into the muscle fiber and are broken up.4 In addition, the complement system is activated to mediate further damage on the postsynaptic membrane.
AChR antibodies may come from germinal centers of the thymus, where clustered myoid cells express AChR on the plasma membrane surface.5 About 60% of AChR antibody-positive myasthenia gravis patients have an enlarged thymus, and 10% have a thymoma—a tumor of the epithelial cells of this organ. Conversely, about 15% of patients with a thymoma have clinical myasthenia gravis, and an additional 20% possess antibodies against AChR in the serum without myasthenic symptoms.5
MuSK antibody-positive myasthenia gravis
Like AChR, MuSK is a transmembrane component of the postsynaptic neuromuscular junction. During formation of the neuromuscular junction, MuSK is activated through the binding of agrin (a nerve-derived proteoglycan) to lipoprotein-related protein 4 (LRP4), after which complicated intracellular signaling promotes the assembly and stabilization of AChR.6
Unlike AChR antibodies, antibodies against MuSK do not activate the complement system, and complement fixation is not essential for clinical myasthenic symptoms to appear.7 Also, myasthenia gravis with MuSK antibodies is rarely associated with thymoma.8
The precise mechanism by which MuSK antibody impairs transmission at the neuromuscular junction has been a mystery until recently. Animal models, including MuSK-mutant mice and mice injected with MuSK protein or with purified immunoglobulin G from patients with this disease, have revealed a significant reduction of AChR clusters and destruction of neuromuscular junction structures.7,9–12
In addition, MuSK antibodies produce pre-synaptic dysfunction, manifesting as a reduction of acetylcholine content. This information is based on studies in mice and on in vitro electrophysiologic analyses of neuromuscular junctions from a patient with this disease.7,9–13
Finally, MuSK antibodies may indirectly affect the recycling of acetylcholine. After post-synaptic activation, acetylcholine is normally hydrolized by acetylcholinesterase, which is located in the synaptic cleft but anchored to MuSK on the postsynaptic membrane. MuSK antibodies block the binding of MuSK to acetylcholinesterase, possibly leading to less accumulation of acetylcholinesterase.14 This process may explain why patients with MuSK antibody-positive myasthenia gravis tend to respond poorly to acetylcholinesterase inhibitors (more about this below).
Seronegative myasthenia gravis
In a series of 562 consecutive patients with generalized weakness due to myasthenia gravis, 92% were positive for AChR antibody, 3% were positive for MuSK antibody, and 5% were seronegative (possessing neither antibody).15 In contrast, about 50% of patients with purely ocular myasthenia gravis (ie, with isolated weakness of the levator palpebrae superioris, orbicularis oculi, or oculomotor muscles) are seropositive for AChR antibody. Only a few ocular MuSK antibody-positive cases have been described, leaving the rest seronegative. Rarely, both antibodies can be detected in the same patient.16
In patients who are negative for AChR antibodies at the time of disease onset, sero-conversion may occur later during the course. Repeating serologic testing 6 to 12 months later may detect AChR antibodies in approximately 15% of patients who were initially seronegative.15,17
The clinical presentation, electrophysiologic findings, thymic pathologic findings, and treatment responses are similar in AChR antibody-positive and seronegative myasthenia gravis.17 Muscle biopsy study in seronegative cases demonstrates a loss of AChR as well.18
Based on these observations, it has been proposed that seronegative patients may have low-affinity antibodies that can bind to tightly clustered AChRs on the postsynaptic membrane but escape detection by routine radioimmunoassays in a solution phase. With a sensitive cell-based immunofluorescence assay, low-affinity antibodies to clustered AChRs were detected in 66% of patients with generalized myasthenia gravis and in 50% of those with ocular myasthenia gravis who were seronegative on standard assays.19,20 These low-affinity AChR antibodies can also activate complement in vitro, increasing the likelihood that they are pathogenic. However, assays to detect low-affinity AChR antibodies are not yet commercially available.
Within the past year, three research groups independently reported detecting antibodies to LRP4 in 2% to 50% of seronegative myasthenia gravis patients. This wide variation in the prevalence of LRP4 antibodies could be related to patient ethnicity and methods of detection.21–23 LRP4 is a receptor for agrin and is required for agrin-induced MuSK activation and AChR clustering. LRP antibodies can activate complement; therefore, it is plausible that LRP4 antibody binding leads to AChR loss on the postsynaptic membrane. However, additional study is needed to determine if LRP4 antibodies are truly pathogenic in myasthenia gravis.
A DISORDER OF FATIGABLE WEAKNESS
Myasthenia gravis is a disorder of fatigable weakness producing fluctuating symptoms. Symptoms related to the involvement of specific muscle groups are listed in Table 1. Muscle weakness is often worse later in the day or after exercise.
Ocular myasthenia gravis accounts for about 15% of all cases. Of patients initially presenting with ocular symptoms only, twothirds will ultimately develop generalized symptoms, most within the first 2 years.24 No factor has been identified that predicts conversion from an ocular to a generalized form.
Several clinical phenotypes of MuSK antibody-positive myasthenia gravis have been described. An oculobulbar form presents with diplopia, ptosis, dysarthria, and profound atrophy of the muscles of the tongue and face. A restricted myopathic form presents with prominent neck, shoulder, and respiratory weakness without ocular involvement. A third form is a combination of ocular and proximal limb weakness, indistinguishable from AChR antibody-positive disease.25
MuSK antibody-positive patients do not respond as well to acetylcholinesterase inhibitors as AChR antibody-positive patients do. In one study, nearly 70% of MuSK antibody-positive patients demonstrated no response, poor tolerance, or cholinergic hypersensitivity to these agents.25 Fortunately, most MuSK antibody-positive patients have a favorable response to immunosuppressive therapy—sometimes a dramatic improvement after plasmapheresis.8
DIAGNOSIS OF MYASTHENIA GRAVIS
The common differential diagnoses for myasthenia gravis are listed in Table 2.
The essential feature of myasthenia gravis is fluctuating muscle weakness, often with fatigue. Many patients complain of weakness of specific muscle groups after their repeated usage. Pain is generally a less conspicuous symptom, and generalized fatigue without objective weakness is inconsistent with myasthenia gravis.
Signs of muscle weakness may include droopy eyelids, diplopia, inability to hold the head straight, difficulty swallowing or chewing, speech disturbances, difficulty breathing, and difficulty raising the arms or rising from the sitting position. A historical pattern of ptosis alternating from one eye to the other is fairly characteristic of myasthenia gravis.
The weakness of orbicularis oculi is easily identified on examination by prying open the eyes during forced eye closure. Limb weakness is usually more significant in the arms than in the legs. An often-neglected feature of myasthenia gravis is finger extensor weakness with a relative sparing of other distal hand muscles.2
The ice-pack test is performed by placing a small bag of ice over the ptotic eye for 2 to 5 minutes and assessing the degree of ptosis for any noticeable improvement. This test is not very helpful for assessing ocular motor weakness.
The edrophonium (Tensilon) test can be used for patients with ptosis or ophthalmoparesis. Edrophonium, a short-acting acetylcholinesterase inhibitor, is given intravenously while the patient is observed for objective improvement. The patient’s cardiovascular status should be monitored for arrhythmias and hypotension. Atropine should be immediately available in case severe bradycardia develops.
The ice-pack test and the edrophonium test can give false-negative and false-positive results, and the diagnosis of myasthenia gravis must be verified by other diagnostic tests.
Testing for antibodies
Testing for circulating AChR antibodies, MuSK antibodies, or both is the first step in the laboratory confirmation of myasthenia gravis.
There are three AChR antibody subtypes: binding, blocking, and modulating. Binding antibodies are present in 80% to 90% of patients with generalized myasthenia gravis and 50% of those with ocular myasthenia gravis. Testing for blocking and modulating AChR antibodies increases the sensitivity by less than 5% when added to testing for binding antibodies.
AChR antibody titers correlate poorly with disease severity between patients. However, in individual patients, antibody titers tend to go down in parallel with clinical improvement.
MuSK antibody is detected in nearly half of myasthenia gravis patients with generalized weakness who are negative for AChR antibody.
Electrophysiologic tests
Electrophysiologic tests can usually confirm the diagnosis of seronegative myasthenia gravis. They are also helpful in seropositive patients who have unusual clinical features or a poor response to treatment.
Repetitive nerve stimulation studies use a slow rate (2–5 Hz) of repetitive electrical stimulation. The study is positive if the motor response declines by more than 10%. However, a decremental response is not specific for myasthenia gravis, as it may be seen in other neuromuscular disorders such as motor neuron disease or Lambert-Eaton myasthenic syndrome.
This test is technically easier to do in distal muscles than in proximal muscles, but less sensitive. Therefore, proximal muscles such as the trapezius or facial muscles are usually also sampled to maximize the yield. To further maximize the sensitivity, muscles being tested should be warm, and acetylcholinesterase inhibitors should be withheld for 12 hours before.
Repetitive nerve stimulation studies in distal muscles are positive in approximately 75% of patients with generalized myasthenia gravis and in 30% with ocular myasthenia gravis.26
Single-fiber electromyography is more technically demanding than repetitive nerve stimulation and is less widely available. It is usually performed with a special needle electrode that can simultaneously identify action potentials arising from individual muscle fibers innervated by the same axon.
Variability in time of the second action potential relative to the first is called “jitter.” Abnormal jitter is seen in more than 95% of patients with generalized myasthenia gravis and in 85% to 90% of those with ocular myasthenia gravis.26,27 However, abnormal jitter can also be seen in other neuromuscular diseases such as motor neuron disease or in neuromuscular junctional disorders such as Lambert-Eaton myasthenic syndrome.
Imaging studies
Chest computed tomography or magnetic resonance imaging with contrast should be performed in all myasthenia gravis patients to look for a thymoma.
TREATMENT OF MYASTHENIA GRAVIS
Acetylcholinesterase inhibitors
As a reasonable first therapy in mild cases of myasthenia gravis, acetylcholinesterase inhibitors slow down the degradation of acetylcholine and prolong its effect in the neuromuscular junction, but they are not disease-modifying and their benefits are mild.
Pyridostigmine is the usual choice of acetylcholinesterase inhibitor. Its onset of action is rapid (15 to 30 minutes) and its action lasts for 3 to 4 hours. For most patients, the effective dosage range is 60 mg to 90 mg every 4 to 6 hours. A long-acting form is also available and can be given as a single nighttime dose.
Immunomodulating therapy
Patients who have moderate to severe symptoms require some form of immunomodulating therapy.
Plasmapheresis or intravenous immune globulin is reserved for patients with severe or rapidly worsening disease because their beneficial effects can be seen within the first week of treatment.
Longer-acting immunotherapies (corticosteroids, azathioprine, mycophenolate mofetil and others) have a slower onset of responses but provide sustained benefits. Which drug to use depends on factors such as comorbidity, side effects, and cost.
Drugs to avoid
A number of medications can exacerbate weakness in myasthenia gravis and should be avoided or used with caution. The list is long, but ones that deserve the most attention are penicillamine, interferons, procainamide, quinidine, and antibiotics, including quinolones and aminoglycosides. A more comprehensive list of medications that may exacerbate myasthenia gravis symptoms can be found in a review by Keesey.2
RAPID INDUCTION IMMUNOTHERAPIES : PLASMAPHERESIS, IMMUNE GLOBULIN
Both plasmapheresis and intravenous immune globulin act quickly over days, but in most patients their effects last only a few weeks. Both are used as rescue therapies for myasthenic crises, bridging therapy to slow-acting immunotherapeutic agents, or maintenance treatment for poorly controlled cases.
Several retrospective studies have confirmed the efficacy of plasmapheresis in more than 80% of patients with generalized symptoms.28,29
In a randomized trial in patients with generalized therapies, intravenous immune globulin improved muscle strength in the group of patients with severe symptoms.30 The effective dosage of intravenous immune globulin varies from 1 to 2 g/kg without observed difference between doses.31 Trials comparing the efficacy of intravenous immune globulin and plasmapheresis in acute and severe myasthenia gravis did not reveal a difference in efficacy.32,33 Intravenous immune globulin at a minimal dose of 0.4 g/kg every 3 months has been successfully used as a long-term maintenance monotherapy, and such a role could be expanded to more patients with further studies.34
The choice between plasmapheresis and intravenous immune globulin is often based on the ability of a patient to tolerate each treatment and on the availability of the plasmapheresis procedure. Intravenous immune globulin is easier to administer, is associated with fewer adverse events related to vascular access, and is therefore more appropriate than plasmapheresis in some centers.
CHRONIC MAINTENANCE IMMUNOMODULATING TREATMENT
Corticosteroids
Prednisone, the most commonly used agent, leads to remission or marked improvement in 70% to 80% of patients with ocular or generalized myasthenia gravis.35 It may also reduce the progression of ocular myasthenia gravis to the generalized form.36
The effective dose of prednisone depends on the severity and distribution of symptoms. Some patients may need up to 1.0 mg/kg/day (usually 50 to 80 mg per day). In patients with mild to moderate symptoms, a lower maximal dosage such as 20 to 40 mg per day can be sufficient.
Within 1 to 2 weeks after starting high-dose prednisone, up to 50% of patients may develop a transient deterioration, including possible precipitation of a myasthenic crisis.37 For this reason, high-dose prednisone is commonly started only in hospitalized patients who are also receiving plasmapheresis or intravenous immune globulin. Otherwise, an outpatient dose-escalation protocol can be used to achieve a target dose over several weeks.
Prednisone tapering can begin after the patient has been on the maximal dose for 1 to 2 months and significant improvement is evident. A monthly tapering of 5 to 10 mg is preferred, then more slowly after the daily dose reaches 30 mg. The usual maintenance dose averages about 5 mg daily.
Common side effects of prednisone include weight gain, cushingoid features, easy bruising, cataracts, glaucoma, hypertension, diabetes, dyslipidemia, and osteoporosis. Patients are advised to take supplemental calcium (1,500 mg per day) and vitamin D (400 to 800 IU per day). For those most at risk of osteoporosis, treatment with a bisphosphonate should be considered.
Other immunotherapeutic agents are often needed, either to replace the corticosteroid or to permit use of lower doses of it. Because of their delayed onset of action, starting such corticosteroid-sparing agents early in the course is often necessary. These agents are often initially combined with high-dose prednisone, with an eventual goal of weaning off prednisone entirely. This strategy offers the advantage of relatively rapid induction while avoiding the long-term adverse effects of corticosteroid treatment.
Azathioprine
Azathioprine doesn’t begin to show a beneficial effect in myasthenia gravis for 6 to 12 months, and it often reaches its maximal efficacy only after 1 to 2 years of treatment.38
In a study of 78 myasthenia gravis patients, 91% improved when treated with azathioprine alone or together with prednisone.39 In another study using azathioprine and prednisolone for generalized myasthenia gravis, nearly two-thirds of patients came off prednisolone while maintaining remission for 3 years.38
A typical maintenance dose is 2 to 3 mg/kg/day. Common side effects are nausea, vomiting, and malaise. Less frequent side effects include hematologic abnormalities, abnormal liver function, and pancreatitis. Monthly monitoring of complete blood cell counts and liver function tests is warranted for the first 6 months, then less often.
One in 300 people in the general population is homozygous for a mutant allele in the thiopurine methyltransferase (TPMT) gene. Patients with this genotype should not receive azathioprine because of the risk of life-threatening bone marrow suppression.40 A slightly increased risk of various forms of lymphoma has been documented.41
Mycophenolate mofetil
A well-tolerated medication with few side effects, mycophenolate mofetil is being used more in myasthenia gravis. The results of two recent randomized trials suggested that it is not effective in improving myasthenia gravis symptoms or sparing prednisone dosage when used for 90 days or 36 weeks.42,43 However, extensive clinical experience supports its longterm efficacy in myasthenia gravis.
In a retrospective study of 85 patients with generalized myasthenia gravis, mycophenolate at doses of 1 to 3 g daily improved symptoms in 73% and produced remission in 50%. Steroid dosage was reduced in 71% of patients.44
Another retrospective study, with 102 patients, verified a slow development of clinical benefit after months of mycophenolate therapy alone or in combination with prednisone. Approximately 50% of patients achieved a minimal manifestation status after 6 to 12 months of mycophenolate treatment. Eventually, at 24 months of treatment, 80% of patients had a desirable outcome of minimal clinical manifestation or better, 55% of patients were able to come off prednisone entirely, and 75% were taking less than 7.5 mg of prednisone per day.45
Common side effects of mycophenolate include nausea, diarrhea, and infections such as urinary tract infections and herpes reactivation. The complete blood cell count needs to be monitored frequently during the first 6 months of therapy. Leukopenia can occur but rarely necessitates stopping mycophenolate. Long-term safety data are lacking, but so far there has been no clearly increased risk of malignancy.
Mycophenolate exposure in pregnancy results in a high incidence of major fetal malformations. Therefore, its use in pregnant patients is discouraged, and women of child-bearing age should use effective contraception.46
Cyclosporine
A randomized trial in a small number of patients suggested that cyclosporine is fairly effective as monotherapy.47 Its onset of action in myasthenia gravis is faster than that of other corticosteroid-sparing agents, and clinical benefit can often be observed as early as 1 to 2 months. A dose of 5 mg/kg/day and a maintenance serum level of 100 to 150 ng/mL are generally recommended. However, renal, hepatic, and hematologic toxicities and interactions with other medications make cyclosporine a less attractive choice.
Methotrexate
A randomized trial evaluated the utility of methotrexate as a steroid-sparing agent compared with azathioprine.48 At 24 months, its steroid-sparing effect was similar to that of azathioprine, and the prednisone dosage had been reduced in more than 50% of patients.
Another phase II trial studying the efficacy of methotrexate in myasthenia gravis is under way.49
Rituximab
Rituximab is a monoclonal antibody against B-cell membrane marker CD20. A growing number of case series support its efficacy in patients with severe generalized myasthenia gravis refractory to multiple immunosuppressants.16,50 It seems particularly effective for MuSK antibody-positive disease, reducing MuSK antibody titers and having a treatment effect that lasts for years.
The standard dosage is 375 mg/m2 per week for 4 consecutive weeks. Peripheral B cells tend to be depleted within 2 weeks after the first infusion, while T-cell populations remain unchanged.50
A minimal infusion reaction such as flushing and chills can be seen with the first infusion. Patients may be more susceptible to certain infections such as reactivation of herpes zoster, but overall rituximab is well tolerated. Rare cases of progressive multifocal leukoencephalopathy have been reported in patients taking it, but none have occurred so far in myasthenia gravis treatment.
Cyclophosphamide
Cyclophosphamide is an alkylating agent that reduces proliferation of both B and T cells. It can be effective in myasthenia gravis, but potentially serious side effects limit its use. It should be reserved for the small percentage of cases that are refractory to other immunotherapies.
Thymectomy
Surgical treatment should be considered for patients with thymoma. If the tumor cannot be surgically resected, chemoradiotherapy can be considered for relief of myasthenic symptoms and for prevention of local invasion.
Thymomas recur in a minority of patients many years after the initial resection, sometimes without myasthenia gravis symptoms. A recurrence of symptoms does not necessarily indicate a recurrence of thymoma. The lack of correlation between myasthenia gravis symptoms and thymoma recurrence highlights the importance of radiologic follow-up in these patients.
For patients without thymoma, many experts believe that thymectomy is beneficial in patients under age 60 who have generalized myasthenia gravis. The likelihood of medication-free remission is about twice as high, and the likelihood of becoming asymptomatic is about one and a half times higher after thymectomy.51 However, it takes up to several years for the benefits of thymectomy to manifest, and thymectomy does not guarantee protection from developing AChR antibody-positive myasthenia gravis in the future.
The optimal timing of thymectomy is not well established; however, the procedure is usually recommended within the first 3 years of diagnosis.52 The response rates from thymectomy are similar for AChR antibody-positive and seronegative patients. In general, thymectomy for MuSK antibody-positive patients has not been effective, and its role in ocular myasthenia gravis is unclear.2,53
Current therapies for myasthenia gravis can help most patients achieve sustained improvement. The overall prognosis has dramatically improved over the last 4 decades: the mortality rate used to be 75%; now it is 4.5%.1
Myasthenia gravis is the most common disorder of neuromuscular junction transmission and is also one of the best characterized autoimmune diseases. However, its symptoms—primarily weakness—vary from patient to patient, and in the same patient, by time of day and over longer time periods. The variation in symptoms can be very confusing to undiagnosed patients and puzzling to unsuspecting physicians. Such diagnostic uncertainty can give the patient additional frustration and emotional stress, which in turn exacerbate his or her condition.
In this review, we will give an overview of the pathogenesis, clinical manifestations, diagnosis, and treatment of myasthenia gravis.
TWO PEAKS IN INCIDENCE BY AGE
The annual incidence of myasthenia gravis is approximately 10 to 20 new cases per million, with a prevalence of about 150 to 200 per million.2
The age of onset has a bimodal distribution, with an early incidence peak in the second to third decade with a female predominance and a late peak in the 6th to the 8th decade with a male predominance.2
Myasthenia gravis is commonly associated with several other autoimmune disorders, including hypothyroidism, hyperthyroidism, systemic lupus erythematosus, rheumatoid arthritis, vitiligo, diabetes, and, more recently recognized, neuromyelitis optica.3
ANTIBODIES AGAINST AChR AND MuSK
In most cases of myasthenia gravis the patient has autoimmune antibodies against constituents of the neuromuscular junction, specifically acetylcholine receptor (AChR) and muscle-specific tyrosine kinase (MuSK) (Figure 1).
AChR antibody-positive myasthenia gravis
When antibodies bind to AChR on the postsynaptic membrane, they cross-link neighboring AChR units, which are absorbed into the muscle fiber and are broken up.4 In addition, the complement system is activated to mediate further damage on the postsynaptic membrane.
AChR antibodies may come from germinal centers of the thymus, where clustered myoid cells express AChR on the plasma membrane surface.5 About 60% of AChR antibody-positive myasthenia gravis patients have an enlarged thymus, and 10% have a thymoma—a tumor of the epithelial cells of this organ. Conversely, about 15% of patients with a thymoma have clinical myasthenia gravis, and an additional 20% possess antibodies against AChR in the serum without myasthenic symptoms.5
MuSK antibody-positive myasthenia gravis
Like AChR, MuSK is a transmembrane component of the postsynaptic neuromuscular junction. During formation of the neuromuscular junction, MuSK is activated through the binding of agrin (a nerve-derived proteoglycan) to lipoprotein-related protein 4 (LRP4), after which complicated intracellular signaling promotes the assembly and stabilization of AChR.6
Unlike AChR antibodies, antibodies against MuSK do not activate the complement system, and complement fixation is not essential for clinical myasthenic symptoms to appear.7 Also, myasthenia gravis with MuSK antibodies is rarely associated with thymoma.8
The precise mechanism by which MuSK antibody impairs transmission at the neuromuscular junction has been a mystery until recently. Animal models, including MuSK-mutant mice and mice injected with MuSK protein or with purified immunoglobulin G from patients with this disease, have revealed a significant reduction of AChR clusters and destruction of neuromuscular junction structures.7,9–12
In addition, MuSK antibodies produce pre-synaptic dysfunction, manifesting as a reduction of acetylcholine content. This information is based on studies in mice and on in vitro electrophysiologic analyses of neuromuscular junctions from a patient with this disease.7,9–13
Finally, MuSK antibodies may indirectly affect the recycling of acetylcholine. After post-synaptic activation, acetylcholine is normally hydrolized by acetylcholinesterase, which is located in the synaptic cleft but anchored to MuSK on the postsynaptic membrane. MuSK antibodies block the binding of MuSK to acetylcholinesterase, possibly leading to less accumulation of acetylcholinesterase.14 This process may explain why patients with MuSK antibody-positive myasthenia gravis tend to respond poorly to acetylcholinesterase inhibitors (more about this below).
Seronegative myasthenia gravis
In a series of 562 consecutive patients with generalized weakness due to myasthenia gravis, 92% were positive for AChR antibody, 3% were positive for MuSK antibody, and 5% were seronegative (possessing neither antibody).15 In contrast, about 50% of patients with purely ocular myasthenia gravis (ie, with isolated weakness of the levator palpebrae superioris, orbicularis oculi, or oculomotor muscles) are seropositive for AChR antibody. Only a few ocular MuSK antibody-positive cases have been described, leaving the rest seronegative. Rarely, both antibodies can be detected in the same patient.16
In patients who are negative for AChR antibodies at the time of disease onset, sero-conversion may occur later during the course. Repeating serologic testing 6 to 12 months later may detect AChR antibodies in approximately 15% of patients who were initially seronegative.15,17
The clinical presentation, electrophysiologic findings, thymic pathologic findings, and treatment responses are similar in AChR antibody-positive and seronegative myasthenia gravis.17 Muscle biopsy study in seronegative cases demonstrates a loss of AChR as well.18
Based on these observations, it has been proposed that seronegative patients may have low-affinity antibodies that can bind to tightly clustered AChRs on the postsynaptic membrane but escape detection by routine radioimmunoassays in a solution phase. With a sensitive cell-based immunofluorescence assay, low-affinity antibodies to clustered AChRs were detected in 66% of patients with generalized myasthenia gravis and in 50% of those with ocular myasthenia gravis who were seronegative on standard assays.19,20 These low-affinity AChR antibodies can also activate complement in vitro, increasing the likelihood that they are pathogenic. However, assays to detect low-affinity AChR antibodies are not yet commercially available.
Within the past year, three research groups independently reported detecting antibodies to LRP4 in 2% to 50% of seronegative myasthenia gravis patients. This wide variation in the prevalence of LRP4 antibodies could be related to patient ethnicity and methods of detection.21–23 LRP4 is a receptor for agrin and is required for agrin-induced MuSK activation and AChR clustering. LRP antibodies can activate complement; therefore, it is plausible that LRP4 antibody binding leads to AChR loss on the postsynaptic membrane. However, additional study is needed to determine if LRP4 antibodies are truly pathogenic in myasthenia gravis.
A DISORDER OF FATIGABLE WEAKNESS
Myasthenia gravis is a disorder of fatigable weakness producing fluctuating symptoms. Symptoms related to the involvement of specific muscle groups are listed in Table 1. Muscle weakness is often worse later in the day or after exercise.
Ocular myasthenia gravis accounts for about 15% of all cases. Of patients initially presenting with ocular symptoms only, twothirds will ultimately develop generalized symptoms, most within the first 2 years.24 No factor has been identified that predicts conversion from an ocular to a generalized form.
Several clinical phenotypes of MuSK antibody-positive myasthenia gravis have been described. An oculobulbar form presents with diplopia, ptosis, dysarthria, and profound atrophy of the muscles of the tongue and face. A restricted myopathic form presents with prominent neck, shoulder, and respiratory weakness without ocular involvement. A third form is a combination of ocular and proximal limb weakness, indistinguishable from AChR antibody-positive disease.25
MuSK antibody-positive patients do not respond as well to acetylcholinesterase inhibitors as AChR antibody-positive patients do. In one study, nearly 70% of MuSK antibody-positive patients demonstrated no response, poor tolerance, or cholinergic hypersensitivity to these agents.25 Fortunately, most MuSK antibody-positive patients have a favorable response to immunosuppressive therapy—sometimes a dramatic improvement after plasmapheresis.8
DIAGNOSIS OF MYASTHENIA GRAVIS
The common differential diagnoses for myasthenia gravis are listed in Table 2.
The essential feature of myasthenia gravis is fluctuating muscle weakness, often with fatigue. Many patients complain of weakness of specific muscle groups after their repeated usage. Pain is generally a less conspicuous symptom, and generalized fatigue without objective weakness is inconsistent with myasthenia gravis.
Signs of muscle weakness may include droopy eyelids, diplopia, inability to hold the head straight, difficulty swallowing or chewing, speech disturbances, difficulty breathing, and difficulty raising the arms or rising from the sitting position. A historical pattern of ptosis alternating from one eye to the other is fairly characteristic of myasthenia gravis.
The weakness of orbicularis oculi is easily identified on examination by prying open the eyes during forced eye closure. Limb weakness is usually more significant in the arms than in the legs. An often-neglected feature of myasthenia gravis is finger extensor weakness with a relative sparing of other distal hand muscles.2
The ice-pack test is performed by placing a small bag of ice over the ptotic eye for 2 to 5 minutes and assessing the degree of ptosis for any noticeable improvement. This test is not very helpful for assessing ocular motor weakness.
The edrophonium (Tensilon) test can be used for patients with ptosis or ophthalmoparesis. Edrophonium, a short-acting acetylcholinesterase inhibitor, is given intravenously while the patient is observed for objective improvement. The patient’s cardiovascular status should be monitored for arrhythmias and hypotension. Atropine should be immediately available in case severe bradycardia develops.
The ice-pack test and the edrophonium test can give false-negative and false-positive results, and the diagnosis of myasthenia gravis must be verified by other diagnostic tests.
Testing for antibodies
Testing for circulating AChR antibodies, MuSK antibodies, or both is the first step in the laboratory confirmation of myasthenia gravis.
There are three AChR antibody subtypes: binding, blocking, and modulating. Binding antibodies are present in 80% to 90% of patients with generalized myasthenia gravis and 50% of those with ocular myasthenia gravis. Testing for blocking and modulating AChR antibodies increases the sensitivity by less than 5% when added to testing for binding antibodies.
AChR antibody titers correlate poorly with disease severity between patients. However, in individual patients, antibody titers tend to go down in parallel with clinical improvement.
MuSK antibody is detected in nearly half of myasthenia gravis patients with generalized weakness who are negative for AChR antibody.
Electrophysiologic tests
Electrophysiologic tests can usually confirm the diagnosis of seronegative myasthenia gravis. They are also helpful in seropositive patients who have unusual clinical features or a poor response to treatment.
Repetitive nerve stimulation studies use a slow rate (2–5 Hz) of repetitive electrical stimulation. The study is positive if the motor response declines by more than 10%. However, a decremental response is not specific for myasthenia gravis, as it may be seen in other neuromuscular disorders such as motor neuron disease or Lambert-Eaton myasthenic syndrome.
This test is technically easier to do in distal muscles than in proximal muscles, but less sensitive. Therefore, proximal muscles such as the trapezius or facial muscles are usually also sampled to maximize the yield. To further maximize the sensitivity, muscles being tested should be warm, and acetylcholinesterase inhibitors should be withheld for 12 hours before.
Repetitive nerve stimulation studies in distal muscles are positive in approximately 75% of patients with generalized myasthenia gravis and in 30% with ocular myasthenia gravis.26
Single-fiber electromyography is more technically demanding than repetitive nerve stimulation and is less widely available. It is usually performed with a special needle electrode that can simultaneously identify action potentials arising from individual muscle fibers innervated by the same axon.
Variability in time of the second action potential relative to the first is called “jitter.” Abnormal jitter is seen in more than 95% of patients with generalized myasthenia gravis and in 85% to 90% of those with ocular myasthenia gravis.26,27 However, abnormal jitter can also be seen in other neuromuscular diseases such as motor neuron disease or in neuromuscular junctional disorders such as Lambert-Eaton myasthenic syndrome.
Imaging studies
Chest computed tomography or magnetic resonance imaging with contrast should be performed in all myasthenia gravis patients to look for a thymoma.
TREATMENT OF MYASTHENIA GRAVIS
Acetylcholinesterase inhibitors
As a reasonable first therapy in mild cases of myasthenia gravis, acetylcholinesterase inhibitors slow down the degradation of acetylcholine and prolong its effect in the neuromuscular junction, but they are not disease-modifying and their benefits are mild.
Pyridostigmine is the usual choice of acetylcholinesterase inhibitor. Its onset of action is rapid (15 to 30 minutes) and its action lasts for 3 to 4 hours. For most patients, the effective dosage range is 60 mg to 90 mg every 4 to 6 hours. A long-acting form is also available and can be given as a single nighttime dose.
Immunomodulating therapy
Patients who have moderate to severe symptoms require some form of immunomodulating therapy.
Plasmapheresis or intravenous immune globulin is reserved for patients with severe or rapidly worsening disease because their beneficial effects can be seen within the first week of treatment.
Longer-acting immunotherapies (corticosteroids, azathioprine, mycophenolate mofetil and others) have a slower onset of responses but provide sustained benefits. Which drug to use depends on factors such as comorbidity, side effects, and cost.
Drugs to avoid
A number of medications can exacerbate weakness in myasthenia gravis and should be avoided or used with caution. The list is long, but ones that deserve the most attention are penicillamine, interferons, procainamide, quinidine, and antibiotics, including quinolones and aminoglycosides. A more comprehensive list of medications that may exacerbate myasthenia gravis symptoms can be found in a review by Keesey.2
RAPID INDUCTION IMMUNOTHERAPIES : PLASMAPHERESIS, IMMUNE GLOBULIN
Both plasmapheresis and intravenous immune globulin act quickly over days, but in most patients their effects last only a few weeks. Both are used as rescue therapies for myasthenic crises, bridging therapy to slow-acting immunotherapeutic agents, or maintenance treatment for poorly controlled cases.
Several retrospective studies have confirmed the efficacy of plasmapheresis in more than 80% of patients with generalized symptoms.28,29
In a randomized trial in patients with generalized therapies, intravenous immune globulin improved muscle strength in the group of patients with severe symptoms.30 The effective dosage of intravenous immune globulin varies from 1 to 2 g/kg without observed difference between doses.31 Trials comparing the efficacy of intravenous immune globulin and plasmapheresis in acute and severe myasthenia gravis did not reveal a difference in efficacy.32,33 Intravenous immune globulin at a minimal dose of 0.4 g/kg every 3 months has been successfully used as a long-term maintenance monotherapy, and such a role could be expanded to more patients with further studies.34
The choice between plasmapheresis and intravenous immune globulin is often based on the ability of a patient to tolerate each treatment and on the availability of the plasmapheresis procedure. Intravenous immune globulin is easier to administer, is associated with fewer adverse events related to vascular access, and is therefore more appropriate than plasmapheresis in some centers.
CHRONIC MAINTENANCE IMMUNOMODULATING TREATMENT
Corticosteroids
Prednisone, the most commonly used agent, leads to remission or marked improvement in 70% to 80% of patients with ocular or generalized myasthenia gravis.35 It may also reduce the progression of ocular myasthenia gravis to the generalized form.36
The effective dose of prednisone depends on the severity and distribution of symptoms. Some patients may need up to 1.0 mg/kg/day (usually 50 to 80 mg per day). In patients with mild to moderate symptoms, a lower maximal dosage such as 20 to 40 mg per day can be sufficient.
Within 1 to 2 weeks after starting high-dose prednisone, up to 50% of patients may develop a transient deterioration, including possible precipitation of a myasthenic crisis.37 For this reason, high-dose prednisone is commonly started only in hospitalized patients who are also receiving plasmapheresis or intravenous immune globulin. Otherwise, an outpatient dose-escalation protocol can be used to achieve a target dose over several weeks.
Prednisone tapering can begin after the patient has been on the maximal dose for 1 to 2 months and significant improvement is evident. A monthly tapering of 5 to 10 mg is preferred, then more slowly after the daily dose reaches 30 mg. The usual maintenance dose averages about 5 mg daily.
Common side effects of prednisone include weight gain, cushingoid features, easy bruising, cataracts, glaucoma, hypertension, diabetes, dyslipidemia, and osteoporosis. Patients are advised to take supplemental calcium (1,500 mg per day) and vitamin D (400 to 800 IU per day). For those most at risk of osteoporosis, treatment with a bisphosphonate should be considered.
Other immunotherapeutic agents are often needed, either to replace the corticosteroid or to permit use of lower doses of it. Because of their delayed onset of action, starting such corticosteroid-sparing agents early in the course is often necessary. These agents are often initially combined with high-dose prednisone, with an eventual goal of weaning off prednisone entirely. This strategy offers the advantage of relatively rapid induction while avoiding the long-term adverse effects of corticosteroid treatment.
Azathioprine
Azathioprine doesn’t begin to show a beneficial effect in myasthenia gravis for 6 to 12 months, and it often reaches its maximal efficacy only after 1 to 2 years of treatment.38
In a study of 78 myasthenia gravis patients, 91% improved when treated with azathioprine alone or together with prednisone.39 In another study using azathioprine and prednisolone for generalized myasthenia gravis, nearly two-thirds of patients came off prednisolone while maintaining remission for 3 years.38
A typical maintenance dose is 2 to 3 mg/kg/day. Common side effects are nausea, vomiting, and malaise. Less frequent side effects include hematologic abnormalities, abnormal liver function, and pancreatitis. Monthly monitoring of complete blood cell counts and liver function tests is warranted for the first 6 months, then less often.
One in 300 people in the general population is homozygous for a mutant allele in the thiopurine methyltransferase (TPMT) gene. Patients with this genotype should not receive azathioprine because of the risk of life-threatening bone marrow suppression.40 A slightly increased risk of various forms of lymphoma has been documented.41
Mycophenolate mofetil
A well-tolerated medication with few side effects, mycophenolate mofetil is being used more in myasthenia gravis. The results of two recent randomized trials suggested that it is not effective in improving myasthenia gravis symptoms or sparing prednisone dosage when used for 90 days or 36 weeks.42,43 However, extensive clinical experience supports its longterm efficacy in myasthenia gravis.
In a retrospective study of 85 patients with generalized myasthenia gravis, mycophenolate at doses of 1 to 3 g daily improved symptoms in 73% and produced remission in 50%. Steroid dosage was reduced in 71% of patients.44
Another retrospective study, with 102 patients, verified a slow development of clinical benefit after months of mycophenolate therapy alone or in combination with prednisone. Approximately 50% of patients achieved a minimal manifestation status after 6 to 12 months of mycophenolate treatment. Eventually, at 24 months of treatment, 80% of patients had a desirable outcome of minimal clinical manifestation or better, 55% of patients were able to come off prednisone entirely, and 75% were taking less than 7.5 mg of prednisone per day.45
Common side effects of mycophenolate include nausea, diarrhea, and infections such as urinary tract infections and herpes reactivation. The complete blood cell count needs to be monitored frequently during the first 6 months of therapy. Leukopenia can occur but rarely necessitates stopping mycophenolate. Long-term safety data are lacking, but so far there has been no clearly increased risk of malignancy.
Mycophenolate exposure in pregnancy results in a high incidence of major fetal malformations. Therefore, its use in pregnant patients is discouraged, and women of child-bearing age should use effective contraception.46
Cyclosporine
A randomized trial in a small number of patients suggested that cyclosporine is fairly effective as monotherapy.47 Its onset of action in myasthenia gravis is faster than that of other corticosteroid-sparing agents, and clinical benefit can often be observed as early as 1 to 2 months. A dose of 5 mg/kg/day and a maintenance serum level of 100 to 150 ng/mL are generally recommended. However, renal, hepatic, and hematologic toxicities and interactions with other medications make cyclosporine a less attractive choice.
Methotrexate
A randomized trial evaluated the utility of methotrexate as a steroid-sparing agent compared with azathioprine.48 At 24 months, its steroid-sparing effect was similar to that of azathioprine, and the prednisone dosage had been reduced in more than 50% of patients.
Another phase II trial studying the efficacy of methotrexate in myasthenia gravis is under way.49
Rituximab
Rituximab is a monoclonal antibody against B-cell membrane marker CD20. A growing number of case series support its efficacy in patients with severe generalized myasthenia gravis refractory to multiple immunosuppressants.16,50 It seems particularly effective for MuSK antibody-positive disease, reducing MuSK antibody titers and having a treatment effect that lasts for years.
The standard dosage is 375 mg/m2 per week for 4 consecutive weeks. Peripheral B cells tend to be depleted within 2 weeks after the first infusion, while T-cell populations remain unchanged.50
A minimal infusion reaction such as flushing and chills can be seen with the first infusion. Patients may be more susceptible to certain infections such as reactivation of herpes zoster, but overall rituximab is well tolerated. Rare cases of progressive multifocal leukoencephalopathy have been reported in patients taking it, but none have occurred so far in myasthenia gravis treatment.
Cyclophosphamide
Cyclophosphamide is an alkylating agent that reduces proliferation of both B and T cells. It can be effective in myasthenia gravis, but potentially serious side effects limit its use. It should be reserved for the small percentage of cases that are refractory to other immunotherapies.
Thymectomy
Surgical treatment should be considered for patients with thymoma. If the tumor cannot be surgically resected, chemoradiotherapy can be considered for relief of myasthenic symptoms and for prevention of local invasion.
Thymomas recur in a minority of patients many years after the initial resection, sometimes without myasthenia gravis symptoms. A recurrence of symptoms does not necessarily indicate a recurrence of thymoma. The lack of correlation between myasthenia gravis symptoms and thymoma recurrence highlights the importance of radiologic follow-up in these patients.
For patients without thymoma, many experts believe that thymectomy is beneficial in patients under age 60 who have generalized myasthenia gravis. The likelihood of medication-free remission is about twice as high, and the likelihood of becoming asymptomatic is about one and a half times higher after thymectomy.51 However, it takes up to several years for the benefits of thymectomy to manifest, and thymectomy does not guarantee protection from developing AChR antibody-positive myasthenia gravis in the future.
The optimal timing of thymectomy is not well established; however, the procedure is usually recommended within the first 3 years of diagnosis.52 The response rates from thymectomy are similar for AChR antibody-positive and seronegative patients. In general, thymectomy for MuSK antibody-positive patients has not been effective, and its role in ocular myasthenia gravis is unclear.2,53
- Alshekhlee A, Miles JD, Katirji B, Preston DC, Kaminski HJ. Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology 2009; 72:1548–1554.
- Keesey JC. Clinical evaluation and management of myasthenia gravis. Muscle Nerve 2004; 29:484–505.
- Leite MI, Coutinho E, Lana-Peixoto M, et al. Myasthenia gravis and neuromyelitis optica spectrum disorder: a multicenter study of 16 patients. Neurology 2012; 78:1601–1607.
- Drachman DB, Angus CW, Adams RN, Michelson JD, Hoffman GJ. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med 1978; 298:1116–1122.
- Fujii Y. The thymus, thymoma and myasthenia gravis. Surg Today 2013; 43:461–466.
- Evoli A, Lindstrom J. Myasthenia gravis with antibodies to MuSK: another step toward solving mystery? Neurology 2011; 77:1783–1784.
- Mori S, Kubo S, Akiyoshi T, et al. Antibodies against muscle-specific kinase impair both presynaptic and postsynaptic functions in a murine model of myasthenia gravis. Am J Pathol 2012; 180:798–810.
- Guptill JT, Sanders DB, Evoli A. Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve 2011; 44:36–40.
- Chevessier F, Girard E, Molgó J, et al. A mouse model for congenital myasthenic syndrome due to MuSK mutations reveals defects in structure and function of neuromuscular junctions. Hum Mol Genet 2008; 17:3577–3595.
- Richman DP, Nishi K, Morell SW, et al. Acute severe animal model of anti-muscle-specific kinase myasthenia: combined postsynaptic and presynaptic changes. Arch Neurol 2012; 69:453–460.
- Klooster R, Plomp JJ, Huijbers MG, et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain 2012; 135:1081–1101.
- Viegas S, Jacobson L, Waters P, et al. Passive and active immunization models of MuSK-Ab positive myasthenia: electrophysiological evidence for pre and postsynaptic defects. Exp Neurol 2012; 234:506–512.
- Niks EH, Kuks JB, Wokke JH, et al. Pre- and postsynaptic neuromuscular junction abnormalities in musk myasthenia. Muscle Nerve 2010; 42:283–288.
- Kawakami Y, Ito M, Hirayama M, et al. Anti-MuSK autoantibodies block binding of collagen Q to MuSK. Neurology 2011; 77:1819–1826.
- Chan KH, Lachance DH, Harper CM, Lennon VA. Frequency of seronegativity in adult-acquired generalized myasthenia gravis. Muscle Nerve 2007; 36:651–658.
- Collongues N, Casez O, Lacour A, et al. Rituximab in refractory and non-refractory myasthenia: a retrospective multicenter study. Muscle Nerve 2012; 46:687–691.
- Sanders DB, Andrews PI, Howard JF, Massey JM. Seronegative myasthenia gravis. Neurology 1997; 48(suppl 5):40S–45S.
- Shiraishi H, Motomura M, Yoshimura T, et al. Acetylcholine receptors loss and postsynaptic damage in MuSK antibody-positive myasthenia gravis. Ann Neurol 2005; 57:289–293.
- Leite MI, Jacob S, Viegas S, et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain 2008; 131:1940–1952.
- Jacob S, Viegas S, Leite MI, et al. Presence and pathogenic relevance of antibodies to clustered acetylcholine receptor in ocular and generalized myasthenia gravis. Arch Neurol 2012; 69:994–1001.
- Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol 2011; 69:418–422.
- Pevzner A, Schoser B, Peters K, et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol 2012; 259:427–435.
- Zhang B, Tzartos JS, Belimezi M, et al. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol 2012; 69:445–451.
- Kupersmith MJ, Latkany R, Homel P. Development of generalized disease at 2 years in patients with ocular myasthenia gravis. Arch Neurol 2003; 60:243–248.
- Pasnoor M, Wolfe GI, Nations S, et al. Clinical findings in MuSK-antibody positive myasthenia gravis: a US experience. Muscle Nerve 2010; 41:370–374.
- Oh SJ, Kim DE, Kuruoglu R, Bradley RJ, Dwyer D. Diagnostic sensitivity of the laboratory tests in myasthenia gravis. Muscle Nerve 1992; 15:720–724.
- Sanders DB, Stålberg EV. AAEM minimonograph #25: single-fiber electromyography. Muscle Nerve 1996; 19:1069–1083.
- Lazo-Langner A, Espinosa-Poblano I, Tirado-Cárdenas N, et al. Therapeutic plasma exchange in Mexico: experience from a single institution. Am J Hematol 2002; 70:16–21.
- Carandina-Maffeis R, Nucci A, Marques JF, et al. Plasmapheresis in the treatment of myasthenia gravis: retrospective study of 26 patients. Arq Neuropsiquiatr 2004; 62:391–395.
- Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology 2007; 68:837–841.
- Gajdos P, Tranchant C, Clair B, et al; Myasthenia Gravis Clinical Study Group. Treatment of myasthenia gravis exacerbation with intravenous immunoglobulin: a randomized double-blind clinical trial. Arch Neurol 2005; 62:1689–1693.
- Rønager J, Ravnborg M, Hermansen I, Vorstrup S. Immunoglobulin treatment versus plasma exchange in patients with chronic moderate to severe myasthenia gravis. Artif Organs 2001; 25:967–973.
- Barth D, Nabavi Nouri M, Ng E, Nwe P, Bril V. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology 2011; 76:2017–2023.
- Wegner B, Ahmed I. Intravenous immunoglobulin monotherapy in long-term treatment of myasthenia gravis. Clin Neurol Neurosurg 2002; 105:3–8.
- Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol 1984; 15:291–298.
- Monsul NT, Patwa HS, Knorr AM, Lesser RL, Goldstein JM. The effect of prednisone on the progression from ocular to generalized myasthenia gravis. J Neurol Sci 2004; 217:131–133.
- Miller RG, Milner-Brown HS, Mirka A. Prednisone-induced worsening of neuromuscular function in myasthenia gravis. Neurology 1986; 36:729–732.
- Palace J, Newsom-Davis J, Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Myasthenia Gravis Study Group. Neurology 1998; 50:1778–1783.
- Mertens HG, Hertel G, Reuther P, Ricker K. Effect of immunosuppressive drugs (azathioprine). Ann N Y Acad Sci 1981; 377:691–699.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Finelli PF. Primary CNS lymphoma in myasthenic on long-term azathioprine. J Neurooncol 2005; 74:91–92.
- Sanders DB, Hart IK, Mantegazza R, et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology 2008; 71:400–406.
- Muscle Study Group. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology 2008; 71:394–399.
- Meriggioli MN, Ciafaloni E, Al-Hayk KA, et al. Mycophenolate mofetil for myasthenia gravis: an analysis of efficacy, safety, and tolerability. Neurology 2003; 61:1438–1440.
- Hehir MK, Burns TM, Alpers J, Conaway MR, Sawa M, Sanders DB. Mycophenolate mofetil in AChR-antibody-positive myasthenia gravis: outcomes in 102 patients. Muscle Nerve 2010; 41:593–598.
- Merlob P, Stahl B, Klinger G. Tetrada of the possible mycophenolate mofetil embryopathy: a review. Reprod Toxicol 2009; 28:105–108.
- Tindall RS, Rollins JA, Phillips JT, Greenlee RG, Wells L, Belendiuk G. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med 1987; 316:719–724.
- Heckmann JM, Rawoot A, Bateman K, Renison R, Badri M. A single-blinded trial of methotrexate versus azathioprine as steroid-sparing agents in generalized myasthenia gravis. BMC Neurol 2011; 11:97.
- Pasnoor M, He J, Herbelin L, Dimachkie M, Barohn RJ; Muscle Study Group. Phase II trial of methotrexate in myasthenia gravis. Ann N Y Acad Sci 2012; 1275:23–28.
- Díaz-Manera J, Martínez-Hernández E, Querol L, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology 2012; 78:189–193.
- Gronseth GS, Barohn RJ. Practice parameter: thymectomy for autoimmune myasthenia gravis (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2000; 55:7–15.
- Kumar V, Kaminski HJ. Treatment of myasthenia gravis. Curr Neurol Neurosci Rep 2011; 11:89–96.
- Pompeo E, Tacconi F, Massa R, Mineo D, Nahmias S, Mineo TC. Long-term outcome of thoracoscopic extended thymectomy for nonthymomatous myasthenia gravis. Eur J Cardiothorac Surg 2009; 36:164–169.
- Alshekhlee A, Miles JD, Katirji B, Preston DC, Kaminski HJ. Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology 2009; 72:1548–1554.
- Keesey JC. Clinical evaluation and management of myasthenia gravis. Muscle Nerve 2004; 29:484–505.
- Leite MI, Coutinho E, Lana-Peixoto M, et al. Myasthenia gravis and neuromyelitis optica spectrum disorder: a multicenter study of 16 patients. Neurology 2012; 78:1601–1607.
- Drachman DB, Angus CW, Adams RN, Michelson JD, Hoffman GJ. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med 1978; 298:1116–1122.
- Fujii Y. The thymus, thymoma and myasthenia gravis. Surg Today 2013; 43:461–466.
- Evoli A, Lindstrom J. Myasthenia gravis with antibodies to MuSK: another step toward solving mystery? Neurology 2011; 77:1783–1784.
- Mori S, Kubo S, Akiyoshi T, et al. Antibodies against muscle-specific kinase impair both presynaptic and postsynaptic functions in a murine model of myasthenia gravis. Am J Pathol 2012; 180:798–810.
- Guptill JT, Sanders DB, Evoli A. Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve 2011; 44:36–40.
- Chevessier F, Girard E, Molgó J, et al. A mouse model for congenital myasthenic syndrome due to MuSK mutations reveals defects in structure and function of neuromuscular junctions. Hum Mol Genet 2008; 17:3577–3595.
- Richman DP, Nishi K, Morell SW, et al. Acute severe animal model of anti-muscle-specific kinase myasthenia: combined postsynaptic and presynaptic changes. Arch Neurol 2012; 69:453–460.
- Klooster R, Plomp JJ, Huijbers MG, et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain 2012; 135:1081–1101.
- Viegas S, Jacobson L, Waters P, et al. Passive and active immunization models of MuSK-Ab positive myasthenia: electrophysiological evidence for pre and postsynaptic defects. Exp Neurol 2012; 234:506–512.
- Niks EH, Kuks JB, Wokke JH, et al. Pre- and postsynaptic neuromuscular junction abnormalities in musk myasthenia. Muscle Nerve 2010; 42:283–288.
- Kawakami Y, Ito M, Hirayama M, et al. Anti-MuSK autoantibodies block binding of collagen Q to MuSK. Neurology 2011; 77:1819–1826.
- Chan KH, Lachance DH, Harper CM, Lennon VA. Frequency of seronegativity in adult-acquired generalized myasthenia gravis. Muscle Nerve 2007; 36:651–658.
- Collongues N, Casez O, Lacour A, et al. Rituximab in refractory and non-refractory myasthenia: a retrospective multicenter study. Muscle Nerve 2012; 46:687–691.
- Sanders DB, Andrews PI, Howard JF, Massey JM. Seronegative myasthenia gravis. Neurology 1997; 48(suppl 5):40S–45S.
- Shiraishi H, Motomura M, Yoshimura T, et al. Acetylcholine receptors loss and postsynaptic damage in MuSK antibody-positive myasthenia gravis. Ann Neurol 2005; 57:289–293.
- Leite MI, Jacob S, Viegas S, et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain 2008; 131:1940–1952.
- Jacob S, Viegas S, Leite MI, et al. Presence and pathogenic relevance of antibodies to clustered acetylcholine receptor in ocular and generalized myasthenia gravis. Arch Neurol 2012; 69:994–1001.
- Higuchi O, Hamuro J, Motomura M, Yamanashi Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann Neurol 2011; 69:418–422.
- Pevzner A, Schoser B, Peters K, et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol 2012; 259:427–435.
- Zhang B, Tzartos JS, Belimezi M, et al. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol 2012; 69:445–451.
- Kupersmith MJ, Latkany R, Homel P. Development of generalized disease at 2 years in patients with ocular myasthenia gravis. Arch Neurol 2003; 60:243–248.
- Pasnoor M, Wolfe GI, Nations S, et al. Clinical findings in MuSK-antibody positive myasthenia gravis: a US experience. Muscle Nerve 2010; 41:370–374.
- Oh SJ, Kim DE, Kuruoglu R, Bradley RJ, Dwyer D. Diagnostic sensitivity of the laboratory tests in myasthenia gravis. Muscle Nerve 1992; 15:720–724.
- Sanders DB, Stålberg EV. AAEM minimonograph #25: single-fiber electromyography. Muscle Nerve 1996; 19:1069–1083.
- Lazo-Langner A, Espinosa-Poblano I, Tirado-Cárdenas N, et al. Therapeutic plasma exchange in Mexico: experience from a single institution. Am J Hematol 2002; 70:16–21.
- Carandina-Maffeis R, Nucci A, Marques JF, et al. Plasmapheresis in the treatment of myasthenia gravis: retrospective study of 26 patients. Arq Neuropsiquiatr 2004; 62:391–395.
- Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology 2007; 68:837–841.
- Gajdos P, Tranchant C, Clair B, et al; Myasthenia Gravis Clinical Study Group. Treatment of myasthenia gravis exacerbation with intravenous immunoglobulin: a randomized double-blind clinical trial. Arch Neurol 2005; 62:1689–1693.
- Rønager J, Ravnborg M, Hermansen I, Vorstrup S. Immunoglobulin treatment versus plasma exchange in patients with chronic moderate to severe myasthenia gravis. Artif Organs 2001; 25:967–973.
- Barth D, Nabavi Nouri M, Ng E, Nwe P, Bril V. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology 2011; 76:2017–2023.
- Wegner B, Ahmed I. Intravenous immunoglobulin monotherapy in long-term treatment of myasthenia gravis. Clin Neurol Neurosurg 2002; 105:3–8.
- Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol 1984; 15:291–298.
- Monsul NT, Patwa HS, Knorr AM, Lesser RL, Goldstein JM. The effect of prednisone on the progression from ocular to generalized myasthenia gravis. J Neurol Sci 2004; 217:131–133.
- Miller RG, Milner-Brown HS, Mirka A. Prednisone-induced worsening of neuromuscular function in myasthenia gravis. Neurology 1986; 36:729–732.
- Palace J, Newsom-Davis J, Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Myasthenia Gravis Study Group. Neurology 1998; 50:1778–1783.
- Mertens HG, Hertel G, Reuther P, Ricker K. Effect of immunosuppressive drugs (azathioprine). Ann N Y Acad Sci 1981; 377:691–699.
- Relling MV, Gardner EE, Sandborn WJ, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther 2011; 89:387–391.
- Finelli PF. Primary CNS lymphoma in myasthenic on long-term azathioprine. J Neurooncol 2005; 74:91–92.
- Sanders DB, Hart IK, Mantegazza R, et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology 2008; 71:400–406.
- Muscle Study Group. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology 2008; 71:394–399.
- Meriggioli MN, Ciafaloni E, Al-Hayk KA, et al. Mycophenolate mofetil for myasthenia gravis: an analysis of efficacy, safety, and tolerability. Neurology 2003; 61:1438–1440.
- Hehir MK, Burns TM, Alpers J, Conaway MR, Sawa M, Sanders DB. Mycophenolate mofetil in AChR-antibody-positive myasthenia gravis: outcomes in 102 patients. Muscle Nerve 2010; 41:593–598.
- Merlob P, Stahl B, Klinger G. Tetrada of the possible mycophenolate mofetil embryopathy: a review. Reprod Toxicol 2009; 28:105–108.
- Tindall RS, Rollins JA, Phillips JT, Greenlee RG, Wells L, Belendiuk G. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med 1987; 316:719–724.
- Heckmann JM, Rawoot A, Bateman K, Renison R, Badri M. A single-blinded trial of methotrexate versus azathioprine as steroid-sparing agents in generalized myasthenia gravis. BMC Neurol 2011; 11:97.
- Pasnoor M, He J, Herbelin L, Dimachkie M, Barohn RJ; Muscle Study Group. Phase II trial of methotrexate in myasthenia gravis. Ann N Y Acad Sci 2012; 1275:23–28.
- Díaz-Manera J, Martínez-Hernández E, Querol L, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology 2012; 78:189–193.
- Gronseth GS, Barohn RJ. Practice parameter: thymectomy for autoimmune myasthenia gravis (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2000; 55:7–15.
- Kumar V, Kaminski HJ. Treatment of myasthenia gravis. Curr Neurol Neurosci Rep 2011; 11:89–96.
- Pompeo E, Tacconi F, Massa R, Mineo D, Nahmias S, Mineo TC. Long-term outcome of thoracoscopic extended thymectomy for nonthymomatous myasthenia gravis. Eur J Cardiothorac Surg 2009; 36:164–169.
KEY POINTS
- In most cases of myasthenia gravis, the patient has antibodies against acetylcholine receptor (AChR) or musclespecific tyrosine kinase (MuSK).
- Myasthenia gravis is diagnosed by clinical signs, bedside tests (the ice-pack test or the edrophonium test), serologic tests for AChR antibodies or MuSK antibodies, and electrophysiologic tests.
- Acetylcholinesterase inhibitors are the first-step therapy, but patients who have moderate to severe symptoms require some form of immunomodulating therapy.
- A number of drugs can exacerbate weakness in myasthenia gravis and should be avoided or used with caution. These include penicillamine, interferons, procainamide, quinidine, and antibiotics such as quinolones and aminoglycosides.