User login
When patients on target-specific oral anticoagulants need surgery
More then 2.5 million patients in the United States are on long-term anticoagulation therapy for atrial fibrillation, venous thromboembolic disease, or mechanical heart valves,1 and the number is expected to rise as the population ages. Each year, about 10% of these patients undergo an invasive procedure or surgery that requires temporary interruption of anticoagulation.2
Most physicians are familiar with the perioperative management of warfarin, a vitamin K antagonist, since for decades it has been the sole oral anticoagulant available. However, many physicians lack experience with the three target-specific oral anticoagulants (TSOACs; also known as “novel” oral anticoagulants) approved so far: the direct thrombin inhibitor dabigatran (Pradaxa) and the direct factor Xa inhibitors rivaroxaban (Xarelto) and apixaban (Eliquis).
With their rapid onset of action, predictable pharmacokinetics, relatively short half-lives, and fewer drug-drug interactions than warfarin, TSOACs overcome many of the limitations of the older oral anticoagulant warfarin. In many ways, these qualities simplify the perioperative management of anticoagulation. At the same time, these new drugs also bring new challenges: caution is needed in patients with renal impairment; the level of anticoagulation is difficult to assess; and there is no specific antidote or standardized procedure to reverse their anticoagulant effect. While various periprocedural protocols for TSOAC therapy have been proposed, evidence-based guidelines are still to come.
This article first discusses the pharmacology of dabigatran, rivaroxaban, and apixaban that is pertinent to the perioperative period. It then briefly reviews the general principles of perioperative management of anticoagulation. The final section provides specific recommendations for the perioperative management of TSOACs.
PHARMACOLOGY OF TARGET-SPECIFIC ORAL ANTICOAGULANTS
Dabigatran, a factor IIa inhibitor
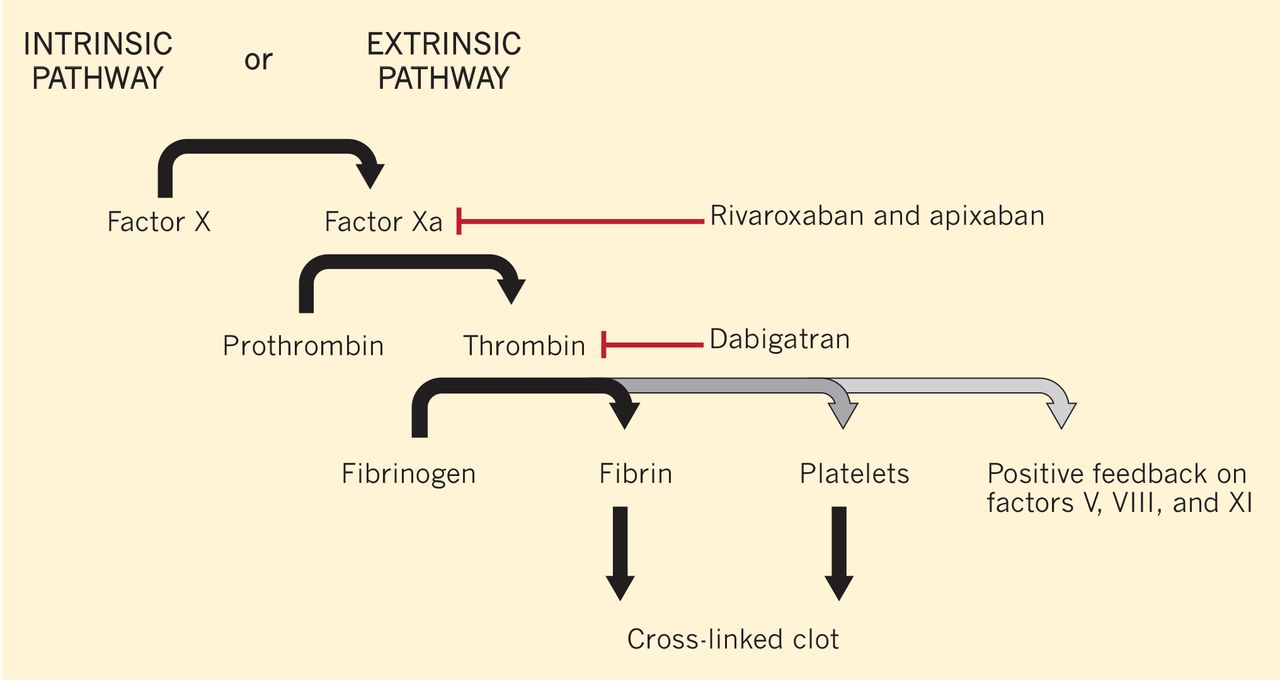
Dabigatran is an oral direct thrombin (factor IIa) inhibitor. It exerts its anticoagulant effect by blocking the generation of fibrin, inhibiting platelet aggregation, and dampening the activity of factors V, VIII, and XI (Figure 1).3,4 From its introduction in October 2010 through August 2012, nearly 3.7 million prescriptions were dispensed to 725,000 patients in the United States.5
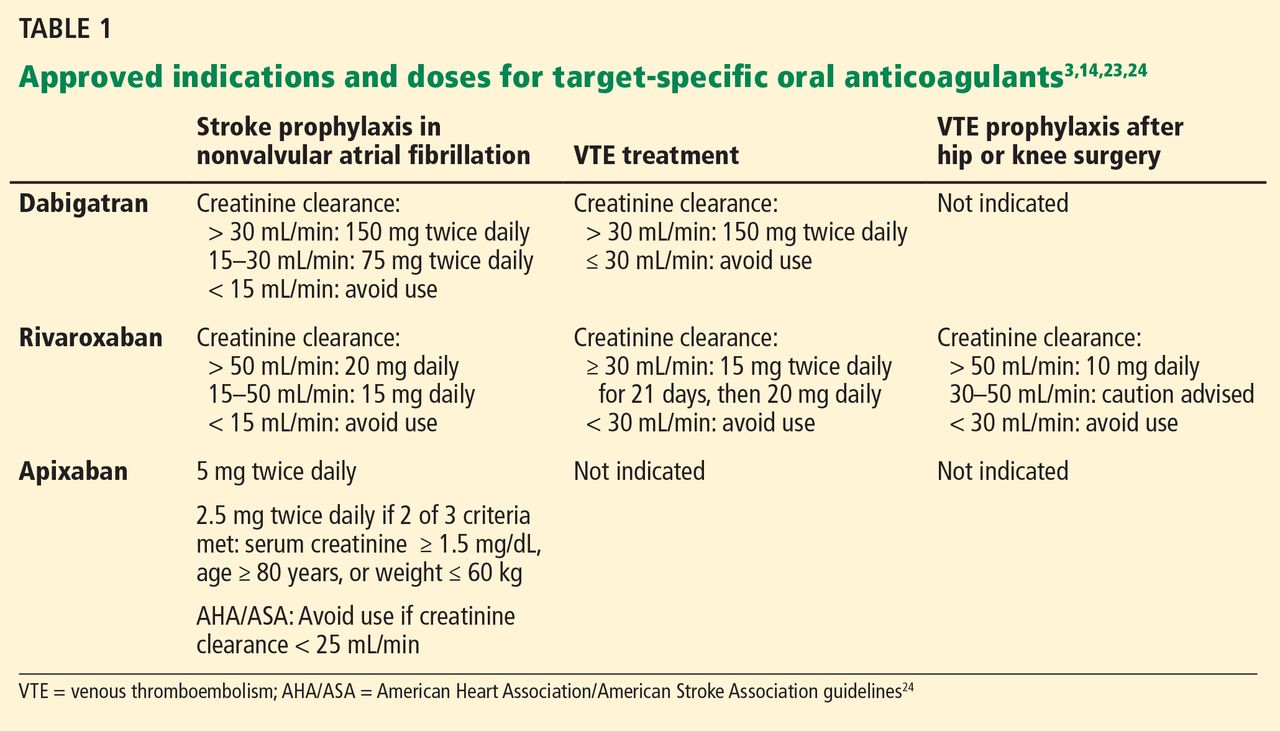
Indications for dabigatran. Dabigatran is approved in the United States and Canada for preventing stroke in nonvalvular atrial fibrillation (Table 1).6 More recently, it received US approval for treating deep vein thrombosis or pulmonary embolism after 5 to 10 days of a parenteral anticoagulant.7,8 It is also approved in Europe and Canada for preventing venous thromboembolism (VTE) after total hip replacement and knee arthroplasty.9,10
Dabigatran is contraindicated in patients with a mechanical heart valve, based on a phase 2 study in which it conferred a higher risk of thromboembolism and bleeding than warfarin.3,11

Pharmacokinetics of dabigatran. Dabigatran is formulated as a prodrug, dabigatran etexilate, in a capsule containing multiple small pellets.12 The capsules should not be crushed, as this significantly increases oral bioavailability. The prodrug is absorbed across the gastric mucosa and is then rapidly converted to the active form (Table 2).
Plasma concentrations peak within 2 hours of ingestion, which means that therapeutic anticoagulation is achieved shortly after taking the drug.
Only 35% of dabigatran is protein-bound, which allows it to be removed by hemodialysis. Nearly 85% of the drug is eliminated in the urine. It has a half-life of 13 to 15 hours in patients with normal renal function.3 However, its half-life increases to about 27 hours in patients whose creatinine clearance is less than 30 mL/min. As a result, the dose must be reduced in patients with renal impairment (Table 1).
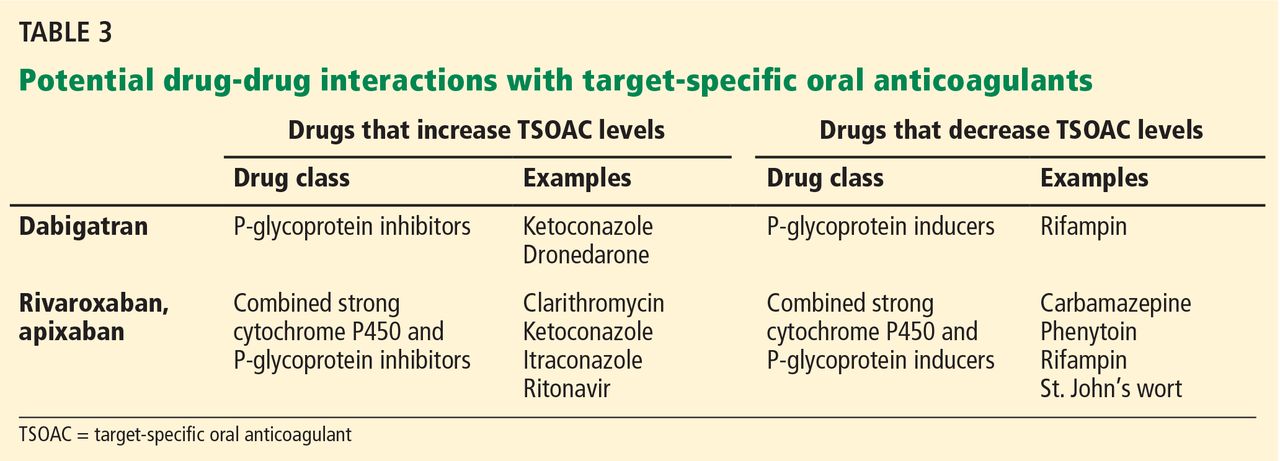
Dabigatran is not metabolized by the cytochrome P450 enzymes, but it is a substrate for P-glycoprotein, so it still has the potential for drug-drug interactions.3 Practitioners should be familiar with these potential interactions (Table 3), as they can result in higher- or lower-than-expected plasma concentrations of dabigatran in the perioperative period.13
Rivaroxaban, a factor Xa inhibitor
Rivaroxaban is an oral direct factor Xa inhibitor. It has been approved by the US Food and Drug Administration (FDA) for the prevention of stroke in nonvalvular atrial fibrillation, for VTE treatment, and for VTE prophylaxis after hip replacement or knee replacement (Table 1).14–20 It has not yet been studied in patients with hip fracture.
Pharmacokinetics of rivaroxaban. Rivaroxaban is manufactured as a tablet that is best absorbed in the stomach (Table 2).14 In contrast to dabigatran, it can be crushed and, for example, mixed with applesauce for patients who have trouble swallowing. It can also be mixed with water and given via nasogastric tube; however, postpyloric administration should be avoided.
Plasma concentrations peak within a few hours after ingestion. Rivaroxaban is highly protein-bound, so it cannot be eliminated by hemodialysis.
The drug relies on renal elimination to a smaller degree than dabigatran, with one-third of the dose eliminated unchanged in the urine, one-third eliminated in the urine as inactive metabolite, and the remaining one-third eliminated in the feces. However, enough parent compound is cleared through the kidneys that the half-life of rivaroxaban increases from 8.3 hours in healthy individuals to 9.5 hours in patients whose creatinine clearance is less than 30 mL/min.21 As with dabigatran, the dose must be adjusted for renal impairment (Table 1).
Rivaroxaban has significant liver metabolism, specifically through the cytochrome P450 3A4 enzyme, and it is also a substrate of P-glycoprotein. Therefore, potential drug-drug interactions must be taken into account, as they may lead to important alterations in plasma concentrations (Table 3).
Apixaban, a factor Xa inhibitor
Apixaban is also an oral direct factor Xa inhibitor. It is the newest of the oral anticoagulants to be approved in the United States, specifically for preventing stroke in nonvalvular atrial fibrillation (Table 1).22
Pharmacokinetics of apixaban. Apixaban is produced as a tablet that is absorbed slowly through the gastrointestinal tract, mainly the distal small bowel and ascending colon (Table 2).23
Peak plasma concentrations are reached a few hours after ingestion. Like rivaroxaban, apixaban is highly protein-bound, so it cannot be removed by hemodialysis.
Apixaban is similar to rivaroxaban in that 27% of the parent compound is cleared through the kidneys, it undergoes significant hepatic metabolism through cytochrome P450 3A4, and it is a substrate for P-glycoprotein.
Drug-drug interactions must be considered as a potential source of altered drug exposure and clearance (Table 3).
Unlike dabigatran and rivaroxaban, dose reduction is not based on the calculated creatinine clearance. Instead, a reduced dose is required if the patient meets two of the following three criteria:
- Serum creatinine level ≥ 1.5 mg/dL
- Age ≥ 80
- Weight ≤ 60 kg (Table 1).
The American Heart Association/American Stroke Association guidelines further recommend against using apixaban in patients with a creatinine clearance less than 25 mL/min.24
Edoxaban, a factor Xa inhibitor in development
Edoxaban (Savaysa), another factor Xa inhibitor, is available in Japan and has been submitted for approval in the United States for treating VTE and for preventing stroke in patients with
PERIOPERATIVE CONSIDERATIONS IN ANTICOAGULATION
Before addressing the perioperative management of TSOACs, let us review the evidence guiding the perioperative management of any chronic anticoagulant.
In fact, no large prospective randomized trial has clearly defined the risks and benefits of using or withholding a bridging anticoagulation strategy around surgery and other procedures, though the PERIOP 2 and BRIDGE trials are currently ongoing.25,26 There are some data regarding continuing anticoagulation without interruption, but they have mainly been derived from specific groups (eg, patients on warfarin undergoing cardiac pacemaker or defibrillator placement) and in procedures that pose a very low risk of bleeding complications (eg, minor dental extractions, cataract surgery, dermatologic procedures).2,27 Recommendations are, therefore, necessarily based on small perioperative trials and data gleaned from cohort review and from studies that did not involve surgical patients.
Ultimately, the decisions whether to discontinue oral anticoagulants and whether to employ bridging anticoagulation are based on assumptions about the risks of bleeding and the risk of thrombotic events, with similar assumptions regarding the effects of anticoagulants on both outcomes. In addition, the relative acceptance of bleeding vs thrombotic risks implicitly guides these complex decisions.
Perioperative bleeding risk
Many risk factors specific to the patient and to the type of surgery affect the rates and severity of perioperative bleeding.28
As for patient-specific risk factors, a small retrospective cohort analysis revealed that a HAS-BLED score of 3 or higher was highly discriminating in predicting perioperative bleeding in atrial fibrillation patients receiving anticoagulation.29 (The HAS-BLED score is based on hypertension, abnormal renal or liver function, stroke, bleeding, labile international normalized ratio [INR], elderly [age > 65] and drug therapy.30) However, there are no widely validated tools that incorporate patient-specific factors to accurately predict bleeding risk in an individual patient.
Therefore, the American College of Chest Physicians (ACCP) guidelines suggest coarsely categorizing bleeding risk as either low or high solely on the basis of the type of procedure.2 Procedures considered “high-risk” have a risk greater than 1.5% to 2% and include urologic surgery involving the prostate or kidney, colonic polyp resections, surgeries involving highly vascular organs such as the liver or spleen, joint replacements, cancer surgeries, and cardiac or neurosurgical procedures.
Perioperative thrombotic risk
The ACCP guidelines2 place patients with atrial fibrillation, VTE, or mechanical heart valves in three risk groups for perioperative thromboembolism without anticoagulation, based on their annual risk of a thrombotic event:
- High risk—annual risk of a thrombotic event > 10%
- Moderate risk—5% to 10%
- Low risk—< 5%.
Comparing the risks calculated by these methods with the real-world risk of perioperative thrombosis highlights the problem of applying nonperioperative risk calculations: the perioperative period exposes patients to a higher risk than these models would predict.31 Nonetheless, these risk categorizations likely have some validity in stratifying patients into risk groups, even if the absolute risks are inaccurate.
Perioperative bridging for patients taking warfarin
Many patients with atrial fibrillation, VTE, or a mechanical heart valve need to interrupt their warfarin therapy because of the bleeding risk of an upcoming procedure.
The perioperative management of warfarin and other vitamin K antagonists is challenging because of the pharmacokinetics and pharmacodynamics of these drugs. Because it has a long half-life, warfarin usually must be stopped 4 to 5 days before a procedure in order to allow not only adequate clearance of the drug itself, but also restoration of functional clotting factors to normal or near-normal levels.12 Warfarin can generally be resumed 12 to 24 hours after surgery, assuming adequate hemostasis has been achieved, and it will again take several days for the INR to reach the therapeutic range.
The ACCP guidelines recommend using the perioperative risk of thromboembolism to make decisions about the need for bridging anticoagulation during warfarin interruption.2 They suggest that patients at high risk of thrombosis receive bridging with an alternative anticoagulant such as low-molecular-weight heparin or unfractionated heparin, because of the prolonged duration of subtherapeutic anticoagulation.
There has been clinical interest in using a TSOAC instead of low-molecular-weight or unfractionated heparin for bridging in the perioperative setting. Although this approach may be attractive from a cost and convenience perspective, it cannot be endorsed as yet because of the lack of information on the pros and cons of such an approach.
Patients at low thrombotic risk do not require bridging. In patients at moderate risk, the decision to bridge or not to bridge is based on careful consideration of patient-specific and surgery-specific factors.
PERIOPERATIVE MANAGEMENT OF TARGET-SPECIFIC ORAL ANTICOAGULANTS
As summarized above, the perioperative management strategy for chronic anticoagulation is based on limited evidence, even for drugs as well established as warfarin.
The most recent ACCP guidelines on the perioperative management of antithrombotic therapy do not mention TSOACs.2 For now, the management strategy must be based on the pharmacokinetics of the drugs, package inserts from the manufacturers, and expert recommendations.3,14,23,32–34 Fortunately, because TSOACs have a more favorable pharmacokinetic profile than that of warfarin, their perioperative uses should be more streamlined. As always, the goal is to minimize the risk of both periprocedural bleeding and thromboembolism.
Timing of cessation of anticoagulation
The timing of cessation of TSOACs before an elective procedure depends primarily on two factors: the bleeding risk of the procedure and the patient’s renal function. Complete clearance of the medication is not necessary in all circumstances.
TSOACs should be stopped four to five half-lives before a procedure with a high bleeding risk, so that there is no or only minimal residual anticoagulant effect. The drug can be stopped two to three half-lives before a procedure with a low bleeding risk. Remember: the half-life increases as creatinine clearance decreases.
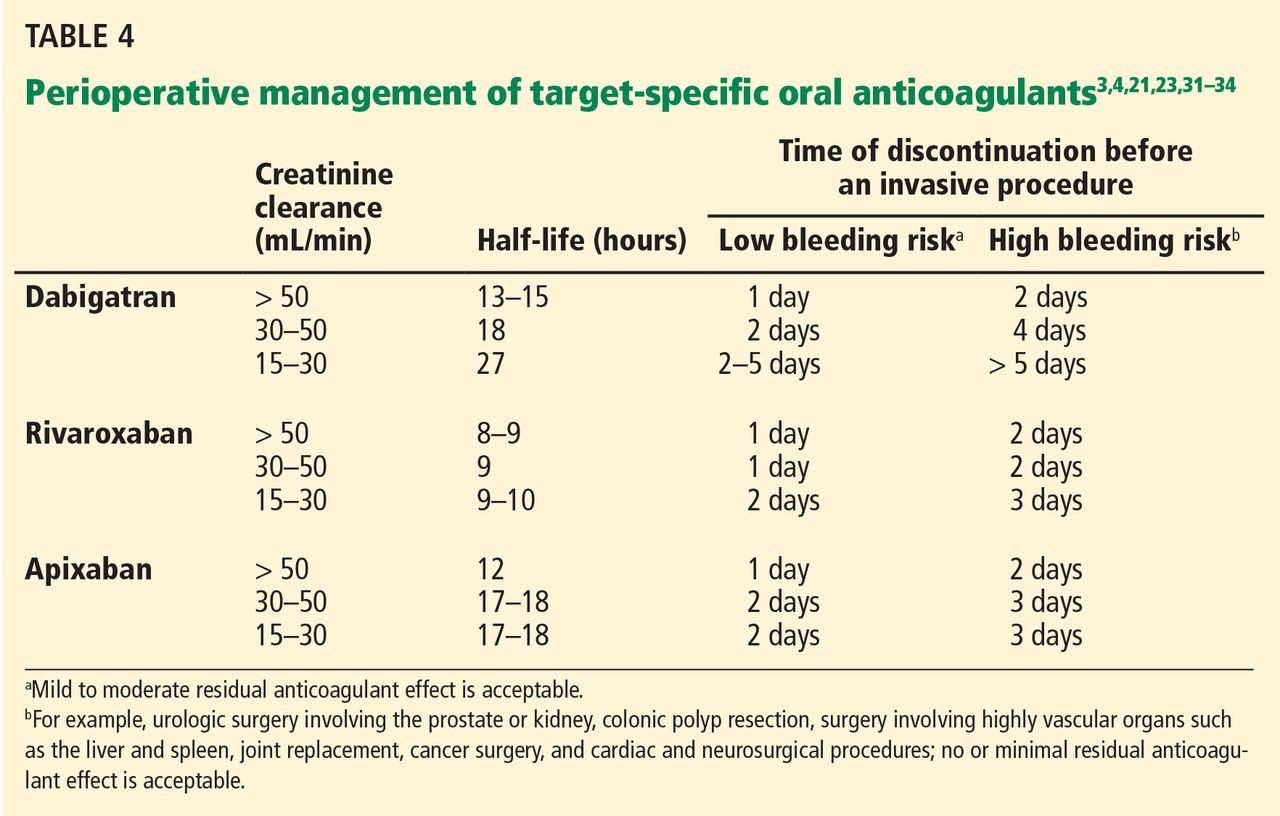
Specific recommendations may vary across institutions, but a suggested strategy is shown in Table 4.3,4,21,23,32–35 For the small subset of patients on P-glycoprotein or cytochrome P450 inhibitors or inducers, further adjustment in the time of discontinuation may be required.
Therapy does not need to be interrupted for procedures with a very low bleeding risk, as defined above.33,34 There is also preliminary evidence that TSOACs, similar to warfarin, may be continued during cardiac pacemaker or defibrillator placement.36
Evidence from clinical trials of perioperative TSOAC management
While the above recommendations are logical, studies are needed to prospectively evaluate perioperative management strategies.
The RE-LY trial (Randomized Evaluation of Long-Term Anticoagulation Therapy), which compared the effects of dabigatran and warfarin in preventing stroke in patients with atrial fibrillation, is one of the few clinical trials that also looked at periprocedural bleeding.37 About a quarter of the RE-LY participants required interruption of anticoagulation for a procedure.
Warfarin was managed according to local practices. For most of the study, the protocol required that dabigatran be discontinued 24 hours before a procedure, regardless of renal function or procedure type. The protocol was later amended and closely mirrored the management plan outlined in Table 4.
With either protocol, there was no statistically significant difference between dabigatran and warfarin in the rates of bleeding and thrombotic complications in the 7 days before or 30 days after the procedure.
A major limitation of the study was that most patients underwent a procedure with a low bleeding risk, so the analysis was likely underpowered to evaluate rates of bleeding in higher-risk procedures.
The ROCKET-AF trial (Rivaroxaban Once-daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation) also shed light on periprocedural bleeding.15 About 15% of the participants required temporary interruption of anticoagulation for a surgical or invasive procedure.38
The study protocol called for discontinuing rivaroxaban 2 days before any procedure. Warfarin was to be held for 4 days to achieve a goal INR of 1.5 or less.15
Rates of major and nonmajor clinically significant bleeding at 30 days were similar with rivaroxaban and with warfarin.38 As with the RE-LY trial, the retrospective analysis was probably underpowered for assessing rates of bleeding in procedures with higher risk.
Perioperative bridging
While stopping a TSOAC in the perioperative period decreases the risk of bleeding, it naturally increases the risk of thromboembolism. However, patients on TSOACs should not routinely require perioperative bridging with an alternative anticoagulant, regardless of thrombotic risk.
Of note, dabigatran, rivaroxaban, and apixaban carry black-box warnings that discontinuation places patients at higher risk of thrombotic events.3,14,23 These warnings further state that coverage with an alternative anticoagulant should be strongly considered during interruption of therapy for reasons other than pathologic bleeding.
However, it does not necessarily follow that perioperative bridging is required. For example, the warning for rivaroxaban is based on the finding in the ROCKET-AF trial that patients in the rivaroxaban group had higher rates of stroke than those in the warfarin group after the study drugs were stopped at the end of the trial.39 While there was initial concern that this could represent a prothrombotic rebound effect, the authors subsequently showed that patients in the rivaroxaban group were more likely to have had a subtherapeutic INR when transitioning to open-label vitamin-K-antagonist therapy.39,40 There was no difference in the rate of stroke or systemic embolism between the rivaroxaban and warfarin groups when anticoagulation was temporarily interrupted for a procedure.38
The risks and benefits of perioperative bridging with TSOACs are difficult to evaluate, given the dearth of trial data. In the RE-LY trial, only 17% of patients on dabigatran and 28% of patients on warfarin underwent periprocedural bridging.37 The selection criteria and protocol for bridging were not reported. In the ROCKET-AF trial, only 9% of patients received bridging therapy despite a mean CHADS2 score of 3.4.38 (The CHADS2 score is calculated as 1 point each for congestive heart failure, hypertension, age ≥ 75, and diabetes; 2 points for stroke or transient ischemic attack.) The decision to bridge or not was left to the individual investigator. As a result, the literature offers diverse opinions about the appropriateness of transitioning to an alternative anticoagulant.41–43
Bridging does not make sense in most instances, since anticoagulants such as low-molecular-weight heparin have pharmacokinetics similar to those of the available TSOACs and also depend on renal clearance.41 However, there may be situations in which patients must be switched to a parenteral anticoagulant such as unfractionated or low-molecular-weight heparin. For example, if a TSOAC has to be held, the patient has acute renal failure, and a needed procedure is still several days away, it would be reasonable to start a heparin drip for an inpatient at increased thrombotic risk.
In patients with normal renal function, these alternative anticoagulants should be started at the time the next TSOAC dose would have been due.3,14,23 In patients with reduced renal function, initiation of an alternative anticoagulant may need to be delayed 12 to 48 hours depending on which TSOAC is being used, as well as on the degree of renal dysfunction. This delay would help ensure that the onset of anticoagulation with the alternative anticoagulant is timed with the offset of therapeutic anticoagulation with the TSOAC.
Although limited, information from available coagulation assays may assist with the timing of initiation of an alternative anticoagulant (see the following section on laboratory monitoring). Serial testing with appropriate coagulation assays may help identify when most of a TSOAC has been cleared from a patient.
Laboratory monitoring
Inevitably, some patients on TSOACs require urgent or emergency surgery. In certain situations, such as before an orthopedic spine procedure, in which the complications of bleeding could be devastating, it may be necessary to know if any residual anticoagulant effect is present.
Monitoring dabigatran. As one might expect, direct thrombin inhibitors such as dabigatran can prolong the prothrombin time and activated partial thromboplastin time (aPTT).44–47 However, the prothrombin time is not recommended for assessing the level of anticoagulation from dabigatran. Many institutions may be using a normal aPTT to rule out therapeutic concentrations of dabigatran, based on results from early in vitro and ex vivo studies.46 While appealing from a practical standpoint, practitioners should exercise caution when relying on the aPTT to assess the risk of perioperative bleeding. A more recent investigation in patients treated with dabigatran found that up to 35% of patients with a normal aPTT still had a plasma concentration in the therapeutic range.48
The thrombin time and ecarin clotting time are more sensitive tests for dabigatran. A normal thrombin time or ecarin clotting time indicates that no or only minimal dabigatran is present.48 Unfortunately, these two tests often are either unavailable or are associated with long turnaround times, which limits their usefulness in the perioperative setting.
Monitoring rivaroxaban and apixaban. Factor Xa inhibitors such as rivaroxaban and apixaban can also influence the prothrombin time and aPTT (Figure 1).44–47,49,50 The aPTT is relatively insensitive to these drugs at low concentrations. It has been suggested that a normal prothrombin time can reasonably exclude therapeutic concentrations of rivaroxaban.45,46 However, the effects on the prothrombin time are highly variable, changing with the reagent used.49,50 In addition, apixaban appears to have less impact on the prothrombin time overall. The INR is not recommended for monitoring the effect of factor Xa inhibitors.
Anti-factor Xa assays likely represent the best option to provide true quantitative information on the level of anticoagulation with either rivaroxaban or apixaban. However, the assays must be specifically calibrated for each drug for results to be useful. (Anti-factor Xa assays cannot be used for heparin or low-molecular-weight heparin.) Further, most institutions do not yet have this capability. When appropriately calibrated, normal anti-factor Xa levels would exclude any effect of rivaroxaban or apixaban.
Reversal of anticoagulation
If patients on TSOACs require emergency surgery or present with significant bleeding in the setting of persistent anticoagulation, it may be necessary to try to reverse the anticoagulation.
Unlike warfarin or heparin, TSOACS do not have specific reversal agents, though specific antidotes are being developed. For example, researchers are evaluating antibodies capable of neutralizing dabigatran, as well as recombinant thrombin and factor Xa molecules that could antagonize dabigatran and rivaroxaban, respectively.51–53
Reversal can be attempted by neutralizing or removing the offending drug. Activated charcoal may be able to reduce absorption of TSOACs that were recently ingested,44 and dabigatran can be removed by hemodialysis.
However, certain practical considerations may limit the use of dialysis in the perioperative period. Insertion of a temporary dialysis line in an anticoagulated patient poses additional bleeding risks. A standard 4-hour hemodialysis session may remove only about 70% of dabigatran from the plasma, which may not be enough to prevent perioperative bleeding.54 Dabigatran also tends to redistribute from adipose tissue back into plasma after each dialysis session.55 Serial sessions of high-flux intermittent hemodialysis or continuous renal replacement therapy may therefore be needed to counteract rebound elevations in the dabigatran concentration.
Reversal can also be attempted through activation of the coagulation cascade via other mechanisms. Fresh-frozen plasma is unlikely to be a practical solution for reversal.44 Although it can readily replace the clotting factors depleted by vitamin K antagonists, large volumes of fresh-frozen plasma would be needed to overwhelm thrombin or factor Xa inhibition by TSOACs.
There are limited data on the use of prothrombin complex concentrates or recombinant activated factor VIIa in patients on TSOACs, though their use can be considered.56 In a trial in 12 healthy participants, a nonactivated four-factor prothrombin complex concentrate containing factors II, VII, IX, and X immediately and completely reversed the anticoagulant effect of rivaroxaban but had no effect on dabigatran.57 Before 2013, there were no nonactivated four-factor prothrombin complex concentrates available in the United States. The FDA has since approved Kcentra for the urgent reversal of vitamin K antagonists, meaning that the reversal of TSOACs in major bleeding events would still be off-label.58 Giving any of the clotting factors carries a risk of thromboembolism.
Resumption of anticoagulation
TSOACs have a rapid onset of action, and therapeutic levels are reached within a few hours of administration.
Extrapolating from the ACCP guidelines, TSOACs can generally be restarted at therapeutic doses 24 hours after low-bleeding-risk procedures.2 Therapeutic dosing should be delayed 48 to 72 hours after a procedure with a high bleeding risk, assuming adequate hemostasis has been achieved. Prophylactic unfractionated heparin or low-molecular-weight heparin therapy can be given in the interim if deemed safe. Alternatively, for orthopedic patients ultimately transitioning back to therapeutic rivaroxaban after hip or knee arthroplasty, prophylactic rivaroxaban doses can be started 6 to 10 hours after surgery.14
There are numerous reasons why the resumption of TSOACs may have to be delayed after surgery, including nothing-by-mouth status, postoperative nausea and vomiting, ileus, gastric or bowel resection, and the anticipated need for future procedures. Since dabigatran capsules cannot be crushed, they cannot be given via nasogastric tube in patients with postoperative dysphagia. Parenteral anticoagulants should be used until these issues resolve.
Unfractionated heparin is still the preferred anticoagulant in unstable or potentially unstable patients, given its ease of monitoring, quick offset of action, and reversibility. When patients have stabilized, TSOACs can be resumed when the next dose of low-molecular-weight heparin would have been due or when the unfractionated heparin drip is discontinued.3,14,23
UNTIL EVIDENCE-BASED GUIDELINES ARE DEVELOPED
The development of TSOACs has ushered in an exciting new era for anticoagulant therapy. Providers involved in perioperative medicine will increasingly encounter patients on dabigatran, rivaroxaban, and apixaban. However, until evidence-based guidelines are developed for these new anticoagulants, clinicians will have to apply their knowledge of pharmacology and critically evaluate expert recommendations in order to manage patients safely throughout the perioperative period.
- Douketis JD, Berger PB, Dunn AS, et al; American College of Chest Physicians. The perioperative management of antithrombotic therapy: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):299S–339S.
- Douketis JD, Spyropoulos AC, Spencer FA, et al; American College of Chest Physicians. Perioperative management of antithrombotic therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e326S–e350S.
- Boehringer Ingelheim Pharmaceuticals, Inc. PRADAXA (dabigatran) package insert. http://bidocs.boehringer-ingelheim.com/BIWebAc-cess/ViewServlet.ser?docBase=renetnt&folderPath=/Prescribing%20Information/PIs/Pradaxa/Pradaxa.pdf. Accessed August 6, 2014.
- Levy JH, Faraoni D, Spring JL, Douketis JD, Samama CM. Managing new oral anticoagulants in the perioperative and intensive care unit setting. Anesthesiology 2013; 118:1466–1474.
- US Food and Drug Administration (FDA). FDA drug safety communication: update on the risk for serious bleeding events with the anticoagulant Pradaxa (dabigatran). www.fda.gov/drugs/drugsafety/ucm326580.htm. Accessed August 6, 2014.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- Schulman S, Kearon C, Kakkar AK, et al; RE-MEDY Trial Investigators. Extended use of dabigatran, warfarin, or placebo in venous thromboembolism. N Engl J Med 2013; 368:709–718.
- Eriksson BI, Dahl OE, Rosencher N, Büller HR, et al; RE-NOVATE Study Group. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement: a randomised, double-blind, non-inferiority trial. Lancet 2007; 370:949–956.
- Eriksson BI, Dahl OE, Rosencher N, et al; RE-MODEL Study Group. Oral dabigatran etexilate vs subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: the RE-MODEL randomized trial. J Thromb Haemost 2007; 5:2178–2185.
- Eikelboom JW, Connolly SJ, Brueckmann M, et al; RE-ALIGN Investigators. Dabigatran versus warfarin in patients with mechanical heart valves. N Engl J Med 2013; 369:1206–1214.
- Ageno W, Gallus AS, Wittkowsky A, Crowther M, Hylek EM, Palareti G; American College of Chest Physicians. Oral anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e44S–e88S.
- Blech S, Ebner T, Ludwig-Schwellinger E, Stangier J, Roth W. The metabolism and disposition of the oral direct thrombin inhibitor, dabigatran, in humans. Drug Metab Dispos 2008; 36:386–399.
- Janssen Pharmaceuticals, Inc. XARELTO (rivaroxaban) package insert. www.xareltohcp.com/about-xarelto/about-xarelto.html?utm_source=google&utm_medium=cpc&utm_campaign=Branded+-+Broad&utm_term=xarelto%20rivaroxaban&utm_content=Xarelto+Rivaroxaban|mkwid|sxSDxPb4m_dc|pcrid|34667840494. Accessed August 6, 2014.
- Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–391.
- EINSTEIN Investigators; Bauersachs R, Berkowitz SD, Brenner B, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med 2010; 363:2499–2510.
- EINSTEIN–PE Investigators; Büller HR, Prins MH, Lensin AW, et al. Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N Engl J Med 2012; 366:1287–1297.
- Eriksson BI, Borris LC, Friedman RJ, et al; RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med 2008; 358:2765–2775.
- Kakkar AK, Brenner B, Dahl OE, et al; RECORD2 Investigators. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet 2008; 372:31–39.
- Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med 2008; 358:2776–2786.
- Kubitza D, Becka M, Mueck W, et al. Effects of renal impairment on the pharmacokinetics, pharmacodynamics and safety of rivaroxaban, an oral, direct factor Xa inhibitor. Br J Clin Pharmacol 2010; 70:703–712.
- Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365:981–992.
- Bristol-Myers Squibb Company. ELIQUIS (apixaban) package insert. www.eliquis.com/index.aspx. Accessed August 6, 2014.
- Furie KL, Goldstein LB, Albers GW, et al; American Heart Association Stroke Council. Oral antithrombotic agents for the prevention of stroke in nonvalvular atrial fibrillation: a science advisory for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:3442–3453.
- ClinicalTrials.gov, US National Institutes of Health. PERIOP 2 - A Safety and Effectiveness Study of LMWH Bridging Therapy Versus Placebo Bridging Therapy for Patients on Long Term Warfarin and Require Temporary Interruption of Their Warfarin. http://clinicaltri-als.gov/show/NCT00432796. Accessed August 6, 2014.
- ClinicalTrials.gov, US National Institutes of Health. Effectiveness of Bridging Anticoagulation for Surgery (The BRIDGE Study). http://clinicaltrials.gov/ct2/show/NCT00786474. Accessed August 6, 2014.
- Birnie DH, Healey JS, Wells GA, et al; BRUISE CONTROL Investigators. Pacemaker or defibrillator surgery without interruption of anticoagulation. N Engl J Med 2013; 368:2084–2093.
- Oberweis BS, Nukala S, Rosenberg A, et al. Thrombotic and bleeding complications after orthopedic surgery. Am Heart J 2013; 165:427.e1–433.e1.
- Omran H, Bauersachs R, Rübenacker S, Goss F, Hammerstingl C. The HAS-BLED score predicts bleedings during bridging of chronic oral anticoagulation. Results from the national multicentre BNK Online bRiDging REgistRy (BORDER). Thromb Haemost 2012; 108:65–73.
- Pisters R, Lane DA, Nieuwlaaat R, de Vos CB, Crijns HJGM, Lip GYH. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation. The Euro Heart Survey. Chest 2010; 138:1093–1100.
- Kaatz S, Douketis JD, Zhou H, Gage BF, White RH. Risk of stroke after surgery in patients with and without chronic atrial fibrillation. J Thromb Haemost 2010; 8:884–890.
- van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost 2010; 103:1116–1127.
- Connolly G, Spyropoulos AC. Practical issues, limitations, and periprocedural management of the NOAC’s. J Thromb Thrombolysis 2013; 36:212–222.
- Spyropoulos AC, Douketis JD. How I treat anticoagulated patients undergoing an elective procedure or surgery. Blood 2012; 120:2954–2962.
- Kaatz S, Kouides PA, Garcia DA, et al. Guidance on the emergent reversal of oral thrombin and factor Xa inhibitors. Am J Hematol 2012; 87(suppl 1):S141–S145.
- Rowley CP, Bernard ML, Brabham WW, et al. Safety of continuous anticoagulation with dabigatran during implantation of cardiac rhythm devices. Am J Cardiol 2013; 111:1165–1168.
- Healey JS, Eikelboom J, Douketis J, et al; RE-LY Investigators. Periprocedural bleeding and thromboembolic events with dabigatran compared with warfarin: results from the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) randomized trial. Circulation 2012; 126:343–348.
- Sherwood MW, Douketis JD, Patel MR, et al; on behalf of the ROCKET AF Investigators. Outcomes of temporary interruption of rivaroxaban compared with warfarin in patients with nonvalvular atrial fibrillation: results from the Rivaroxaban Once Daily, Oral, Direct Factor Xa Inhibition Compared with Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation (ROCKET AF). Circulation 2014; 129:1850–1859.
- Patel MR, Hellkamp AS, Lokhnygina Y, et al. Outcomes of discontinuing rivaroxaban compared with warfarin in patients with nonvalvular atrial fibrillation: analysis from the ROCKET AF trial (Rivaroxaban Once-Daily, Oral, Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation). J Am Coll Cardiol 2013; 61:651–658.
- Reynolds MR. Discontinuation of rivaroxaban: filling in the gaps. J Am Coll Cardiol 2013; 61:659–660.
- Turpie AG, Kreutz R, Llau J, Norrving B, Haas S. Management consensus guidance for the use of rivaroxaban—an oral, direct factor Xa inhibitor. Thromb Haemost 2012; 108:876–886.
- Gallego P, Apostolakis S, Lip GY. Bridging evidence-based practice and practice-based evidence in periprocedural anticoagulation. Circulation 2012; 126:1573–1576.
- Sié P, Samama CM, Godier A, et al; Working Group on Perioperative Haemostasis. Surgery and invasive procedures in patients on long-term treatment with direct oral anticoagulants: thrombin or factor-Xa inhibitors. Recommendations of the Working Group on Perioperative Haemostasis and the French Study Group on Thrombosis and Haemostasis. Arch Cardiovasc Dis 2011; 104:669–676.
- King CS, Holley AB, Moores LK. Moving toward a more ideal anticoagulant: the oral direct thrombin and factor Xa inhibitors. Chest 2013; 143:1106–1116.
- Baglin T, Hillarp A, Tripodi A, Elalamy I, Buller H, Ageno W. Measuring oral direct inhibitors (ODIs) of thrombin and factor Xa: a recommendation from the Subcommittee on Control of Anticoagulation of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2013; 11:756–760.
- Baglin T, Keeling D, Kitchen S; British Committee for Standards in Haematology. Effects on routine coagulation screens and assessment of anticoagulant intensity in patients taking oral dabigatran or rivaroxaban: guidance from the British Committee for Standards in Haematology. Br J Haematol 2012; 159:427–429.
- Mani H, Kasper A, Lindhoff-Last E. Measuring the anticoagulant effects of target specific oral anticoagulants—reasons, methods and current limitations. J Thromb Thrombolysis 2013; 36:187–194.
- Hawes EM, Deal AM, Funk-Adcock D, et al. Performance of coagulation tests in patients on therapeutic doses of dabigatran: a cross-sectional pharmacodynamic study based on peak and trough plasma levels. J Thromb Haemost 2013; 11:1493–1502.
- Barrett YC, Wang Z, Frost C, Shenker A. Clinical laboratory measurement of direct factor Xa inhibitors: anti-Xa assay is preferable to prothrombin time assay. Thromb Haemost 2010; 104:1263–1271.
- Smythe MA, Fanikos J, Gulseth MP, et al. Rivaroxaban: practical considerations for ensuring safety and efficacy. Pharmacotherapy 2013; 33:1223–1245.
- Van Ryn J, Litzenburger T, Waterman A, et al. Dabigatran anticoagulant activity is neutralized by an antibody selective to dabigatran in in vitro and in vivo models. J Am Coll Cardiol 2011; 57:E1130.
- Sheffield W, Lambourne M, Bhakta V, Eltringham-Smith L, Arnold D, Crowther M. Active site-mutated thrombin S195A but not active site-blocked thrombin counteracts the anticoagulant activity of dabigatran in plasma. Abstract presented at the International Society of Thrombosis and Haemostasis 2013 Congress. http://onlinelibrary.wiley.com/doi/10.1111/jth.2013.11.issue-s2/issuetoc. Accessed August 6, 2014.
- Lu G, Luan P, Hollenbach SJ, et al. Reconstructed recombinant factor Xa as an antidote to reverse anticoagulation by factor Xa inhibitors (abstract). J Thromb Haemost 2009; 7(suppl 2):abstract OC-TH-107.
- Stangier J, Rathgen K, Stähle H, Mazur D. Influence of renal impairment on the pharmacokinetics and pharmacodynamics of oral dabigatran etexilate: an open-label, parallel-group, single-centre study. Clin Pharmacokinet 2010; 49:259–268.
- Singh T, Maw TT, Henry BL, et al. Extracorporeal therapy for dabigatran removal in the treatment of acute bleeding: a single center experience. Clin J Am Soc Nephrol 2013; 8:1533–1539.
- Kaatz S, Crowther M. Reversal of target-specific oral anticoagulants. J Thromb Thrombolysis 2013; 36:195–202.
- Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation 2011; 124:1573–1579.
- Sarode R, Milling TJ, Refaai MA, et al. Efficacy and safety of a 4-factor prothrombin complex concentrate in patients on vitamin K antagonists presenting with major bleeding: a randomized, plasma-controlled, phase IIIb study. Circulation 2013; 128:1234–1243.
More then 2.5 million patients in the United States are on long-term anticoagulation therapy for atrial fibrillation, venous thromboembolic disease, or mechanical heart valves,1 and the number is expected to rise as the population ages. Each year, about 10% of these patients undergo an invasive procedure or surgery that requires temporary interruption of anticoagulation.2
Most physicians are familiar with the perioperative management of warfarin, a vitamin K antagonist, since for decades it has been the sole oral anticoagulant available. However, many physicians lack experience with the three target-specific oral anticoagulants (TSOACs; also known as “novel” oral anticoagulants) approved so far: the direct thrombin inhibitor dabigatran (Pradaxa) and the direct factor Xa inhibitors rivaroxaban (Xarelto) and apixaban (Eliquis).
With their rapid onset of action, predictable pharmacokinetics, relatively short half-lives, and fewer drug-drug interactions than warfarin, TSOACs overcome many of the limitations of the older oral anticoagulant warfarin. In many ways, these qualities simplify the perioperative management of anticoagulation. At the same time, these new drugs also bring new challenges: caution is needed in patients with renal impairment; the level of anticoagulation is difficult to assess; and there is no specific antidote or standardized procedure to reverse their anticoagulant effect. While various periprocedural protocols for TSOAC therapy have been proposed, evidence-based guidelines are still to come.
This article first discusses the pharmacology of dabigatran, rivaroxaban, and apixaban that is pertinent to the perioperative period. It then briefly reviews the general principles of perioperative management of anticoagulation. The final section provides specific recommendations for the perioperative management of TSOACs.
PHARMACOLOGY OF TARGET-SPECIFIC ORAL ANTICOAGULANTS
Dabigatran, a factor IIa inhibitor
Dabigatran is an oral direct thrombin (factor IIa) inhibitor. It exerts its anticoagulant effect by blocking the generation of fibrin, inhibiting platelet aggregation, and dampening the activity of factors V, VIII, and XI (Figure 1).3,4 From its introduction in October 2010 through August 2012, nearly 3.7 million prescriptions were dispensed to 725,000 patients in the United States.5
Indications for dabigatran. Dabigatran is approved in the United States and Canada for preventing stroke in nonvalvular atrial fibrillation (Table 1).6 More recently, it received US approval for treating deep vein thrombosis or pulmonary embolism after 5 to 10 days of a parenteral anticoagulant.7,8 It is also approved in Europe and Canada for preventing venous thromboembolism (VTE) after total hip replacement and knee arthroplasty.9,10
Dabigatran is contraindicated in patients with a mechanical heart valve, based on a phase 2 study in which it conferred a higher risk of thromboembolism and bleeding than warfarin.3,11
Pharmacokinetics of dabigatran. Dabigatran is formulated as a prodrug, dabigatran etexilate, in a capsule containing multiple small pellets.12 The capsules should not be crushed, as this significantly increases oral bioavailability. The prodrug is absorbed across the gastric mucosa and is then rapidly converted to the active form (Table 2).
Plasma concentrations peak within 2 hours of ingestion, which means that therapeutic anticoagulation is achieved shortly after taking the drug.
Only 35% of dabigatran is protein-bound, which allows it to be removed by hemodialysis. Nearly 85% of the drug is eliminated in the urine. It has a half-life of 13 to 15 hours in patients with normal renal function.3 However, its half-life increases to about 27 hours in patients whose creatinine clearance is less than 30 mL/min. As a result, the dose must be reduced in patients with renal impairment (Table 1).
Dabigatran is not metabolized by the cytochrome P450 enzymes, but it is a substrate for P-glycoprotein, so it still has the potential for drug-drug interactions.3 Practitioners should be familiar with these potential interactions (Table 3), as they can result in higher- or lower-than-expected plasma concentrations of dabigatran in the perioperative period.13
Rivaroxaban, a factor Xa inhibitor
Rivaroxaban is an oral direct factor Xa inhibitor. It has been approved by the US Food and Drug Administration (FDA) for the prevention of stroke in nonvalvular atrial fibrillation, for VTE treatment, and for VTE prophylaxis after hip replacement or knee replacement (Table 1).14–20 It has not yet been studied in patients with hip fracture.
Pharmacokinetics of rivaroxaban. Rivaroxaban is manufactured as a tablet that is best absorbed in the stomach (Table 2).14 In contrast to dabigatran, it can be crushed and, for example, mixed with applesauce for patients who have trouble swallowing. It can also be mixed with water and given via nasogastric tube; however, postpyloric administration should be avoided.
Plasma concentrations peak within a few hours after ingestion. Rivaroxaban is highly protein-bound, so it cannot be eliminated by hemodialysis.
The drug relies on renal elimination to a smaller degree than dabigatran, with one-third of the dose eliminated unchanged in the urine, one-third eliminated in the urine as inactive metabolite, and the remaining one-third eliminated in the feces. However, enough parent compound is cleared through the kidneys that the half-life of rivaroxaban increases from 8.3 hours in healthy individuals to 9.5 hours in patients whose creatinine clearance is less than 30 mL/min.21 As with dabigatran, the dose must be adjusted for renal impairment (Table 1).
Rivaroxaban has significant liver metabolism, specifically through the cytochrome P450 3A4 enzyme, and it is also a substrate of P-glycoprotein. Therefore, potential drug-drug interactions must be taken into account, as they may lead to important alterations in plasma concentrations (Table 3).
Apixaban, a factor Xa inhibitor
Apixaban is also an oral direct factor Xa inhibitor. It is the newest of the oral anticoagulants to be approved in the United States, specifically for preventing stroke in nonvalvular atrial fibrillation (Table 1).22
Pharmacokinetics of apixaban. Apixaban is produced as a tablet that is absorbed slowly through the gastrointestinal tract, mainly the distal small bowel and ascending colon (Table 2).23
Peak plasma concentrations are reached a few hours after ingestion. Like rivaroxaban, apixaban is highly protein-bound, so it cannot be removed by hemodialysis.
Apixaban is similar to rivaroxaban in that 27% of the parent compound is cleared through the kidneys, it undergoes significant hepatic metabolism through cytochrome P450 3A4, and it is a substrate for P-glycoprotein.
Drug-drug interactions must be considered as a potential source of altered drug exposure and clearance (Table 3).
Unlike dabigatran and rivaroxaban, dose reduction is not based on the calculated creatinine clearance. Instead, a reduced dose is required if the patient meets two of the following three criteria:
- Serum creatinine level ≥ 1.5 mg/dL
- Age ≥ 80
- Weight ≤ 60 kg (Table 1).
The American Heart Association/American Stroke Association guidelines further recommend against using apixaban in patients with a creatinine clearance less than 25 mL/min.24
Edoxaban, a factor Xa inhibitor in development
Edoxaban (Savaysa), another factor Xa inhibitor, is available in Japan and has been submitted for approval in the United States for treating VTE and for preventing stroke in patients with
PERIOPERATIVE CONSIDERATIONS IN ANTICOAGULATION
Before addressing the perioperative management of TSOACs, let us review the evidence guiding the perioperative management of any chronic anticoagulant.
In fact, no large prospective randomized trial has clearly defined the risks and benefits of using or withholding a bridging anticoagulation strategy around surgery and other procedures, though the PERIOP 2 and BRIDGE trials are currently ongoing.25,26 There are some data regarding continuing anticoagulation without interruption, but they have mainly been derived from specific groups (eg, patients on warfarin undergoing cardiac pacemaker or defibrillator placement) and in procedures that pose a very low risk of bleeding complications (eg, minor dental extractions, cataract surgery, dermatologic procedures).2,27 Recommendations are, therefore, necessarily based on small perioperative trials and data gleaned from cohort review and from studies that did not involve surgical patients.
Ultimately, the decisions whether to discontinue oral anticoagulants and whether to employ bridging anticoagulation are based on assumptions about the risks of bleeding and the risk of thrombotic events, with similar assumptions regarding the effects of anticoagulants on both outcomes. In addition, the relative acceptance of bleeding vs thrombotic risks implicitly guides these complex decisions.
Perioperative bleeding risk
Many risk factors specific to the patient and to the type of surgery affect the rates and severity of perioperative bleeding.28
As for patient-specific risk factors, a small retrospective cohort analysis revealed that a HAS-BLED score of 3 or higher was highly discriminating in predicting perioperative bleeding in atrial fibrillation patients receiving anticoagulation.29 (The HAS-BLED score is based on hypertension, abnormal renal or liver function, stroke, bleeding, labile international normalized ratio [INR], elderly [age > 65] and drug therapy.30) However, there are no widely validated tools that incorporate patient-specific factors to accurately predict bleeding risk in an individual patient.
Therefore, the American College of Chest Physicians (ACCP) guidelines suggest coarsely categorizing bleeding risk as either low or high solely on the basis of the type of procedure.2 Procedures considered “high-risk” have a risk greater than 1.5% to 2% and include urologic surgery involving the prostate or kidney, colonic polyp resections, surgeries involving highly vascular organs such as the liver or spleen, joint replacements, cancer surgeries, and cardiac or neurosurgical procedures.
Perioperative thrombotic risk
The ACCP guidelines2 place patients with atrial fibrillation, VTE, or mechanical heart valves in three risk groups for perioperative thromboembolism without anticoagulation, based on their annual risk of a thrombotic event:
- High risk—annual risk of a thrombotic event > 10%
- Moderate risk—5% to 10%
- Low risk—< 5%.
Comparing the risks calculated by these methods with the real-world risk of perioperative thrombosis highlights the problem of applying nonperioperative risk calculations: the perioperative period exposes patients to a higher risk than these models would predict.31 Nonetheless, these risk categorizations likely have some validity in stratifying patients into risk groups, even if the absolute risks are inaccurate.
Perioperative bridging for patients taking warfarin
Many patients with atrial fibrillation, VTE, or a mechanical heart valve need to interrupt their warfarin therapy because of the bleeding risk of an upcoming procedure.
The perioperative management of warfarin and other vitamin K antagonists is challenging because of the pharmacokinetics and pharmacodynamics of these drugs. Because it has a long half-life, warfarin usually must be stopped 4 to 5 days before a procedure in order to allow not only adequate clearance of the drug itself, but also restoration of functional clotting factors to normal or near-normal levels.12 Warfarin can generally be resumed 12 to 24 hours after surgery, assuming adequate hemostasis has been achieved, and it will again take several days for the INR to reach the therapeutic range.
The ACCP guidelines recommend using the perioperative risk of thromboembolism to make decisions about the need for bridging anticoagulation during warfarin interruption.2 They suggest that patients at high risk of thrombosis receive bridging with an alternative anticoagulant such as low-molecular-weight heparin or unfractionated heparin, because of the prolonged duration of subtherapeutic anticoagulation.
There has been clinical interest in using a TSOAC instead of low-molecular-weight or unfractionated heparin for bridging in the perioperative setting. Although this approach may be attractive from a cost and convenience perspective, it cannot be endorsed as yet because of the lack of information on the pros and cons of such an approach.
Patients at low thrombotic risk do not require bridging. In patients at moderate risk, the decision to bridge or not to bridge is based on careful consideration of patient-specific and surgery-specific factors.
PERIOPERATIVE MANAGEMENT OF TARGET-SPECIFIC ORAL ANTICOAGULANTS
As summarized above, the perioperative management strategy for chronic anticoagulation is based on limited evidence, even for drugs as well established as warfarin.
The most recent ACCP guidelines on the perioperative management of antithrombotic therapy do not mention TSOACs.2 For now, the management strategy must be based on the pharmacokinetics of the drugs, package inserts from the manufacturers, and expert recommendations.3,14,23,32–34 Fortunately, because TSOACs have a more favorable pharmacokinetic profile than that of warfarin, their perioperative uses should be more streamlined. As always, the goal is to minimize the risk of both periprocedural bleeding and thromboembolism.
Timing of cessation of anticoagulation
The timing of cessation of TSOACs before an elective procedure depends primarily on two factors: the bleeding risk of the procedure and the patient’s renal function. Complete clearance of the medication is not necessary in all circumstances.
TSOACs should be stopped four to five half-lives before a procedure with a high bleeding risk, so that there is no or only minimal residual anticoagulant effect. The drug can be stopped two to three half-lives before a procedure with a low bleeding risk. Remember: the half-life increases as creatinine clearance decreases.
Specific recommendations may vary across institutions, but a suggested strategy is shown in Table 4.3,4,21,23,32–35 For the small subset of patients on P-glycoprotein or cytochrome P450 inhibitors or inducers, further adjustment in the time of discontinuation may be required.
Therapy does not need to be interrupted for procedures with a very low bleeding risk, as defined above.33,34 There is also preliminary evidence that TSOACs, similar to warfarin, may be continued during cardiac pacemaker or defibrillator placement.36
Evidence from clinical trials of perioperative TSOAC management
While the above recommendations are logical, studies are needed to prospectively evaluate perioperative management strategies.
The RE-LY trial (Randomized Evaluation of Long-Term Anticoagulation Therapy), which compared the effects of dabigatran and warfarin in preventing stroke in patients with atrial fibrillation, is one of the few clinical trials that also looked at periprocedural bleeding.37 About a quarter of the RE-LY participants required interruption of anticoagulation for a procedure.
Warfarin was managed according to local practices. For most of the study, the protocol required that dabigatran be discontinued 24 hours before a procedure, regardless of renal function or procedure type. The protocol was later amended and closely mirrored the management plan outlined in Table 4.
With either protocol, there was no statistically significant difference between dabigatran and warfarin in the rates of bleeding and thrombotic complications in the 7 days before or 30 days after the procedure.
A major limitation of the study was that most patients underwent a procedure with a low bleeding risk, so the analysis was likely underpowered to evaluate rates of bleeding in higher-risk procedures.
The ROCKET-AF trial (Rivaroxaban Once-daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation) also shed light on periprocedural bleeding.15 About 15% of the participants required temporary interruption of anticoagulation for a surgical or invasive procedure.38
The study protocol called for discontinuing rivaroxaban 2 days before any procedure. Warfarin was to be held for 4 days to achieve a goal INR of 1.5 or less.15
Rates of major and nonmajor clinically significant bleeding at 30 days were similar with rivaroxaban and with warfarin.38 As with the RE-LY trial, the retrospective analysis was probably underpowered for assessing rates of bleeding in procedures with higher risk.
Perioperative bridging
While stopping a TSOAC in the perioperative period decreases the risk of bleeding, it naturally increases the risk of thromboembolism. However, patients on TSOACs should not routinely require perioperative bridging with an alternative anticoagulant, regardless of thrombotic risk.
Of note, dabigatran, rivaroxaban, and apixaban carry black-box warnings that discontinuation places patients at higher risk of thrombotic events.3,14,23 These warnings further state that coverage with an alternative anticoagulant should be strongly considered during interruption of therapy for reasons other than pathologic bleeding.
However, it does not necessarily follow that perioperative bridging is required. For example, the warning for rivaroxaban is based on the finding in the ROCKET-AF trial that patients in the rivaroxaban group had higher rates of stroke than those in the warfarin group after the study drugs were stopped at the end of the trial.39 While there was initial concern that this could represent a prothrombotic rebound effect, the authors subsequently showed that patients in the rivaroxaban group were more likely to have had a subtherapeutic INR when transitioning to open-label vitamin-K-antagonist therapy.39,40 There was no difference in the rate of stroke or systemic embolism between the rivaroxaban and warfarin groups when anticoagulation was temporarily interrupted for a procedure.38
The risks and benefits of perioperative bridging with TSOACs are difficult to evaluate, given the dearth of trial data. In the RE-LY trial, only 17% of patients on dabigatran and 28% of patients on warfarin underwent periprocedural bridging.37 The selection criteria and protocol for bridging were not reported. In the ROCKET-AF trial, only 9% of patients received bridging therapy despite a mean CHADS2 score of 3.4.38 (The CHADS2 score is calculated as 1 point each for congestive heart failure, hypertension, age ≥ 75, and diabetes; 2 points for stroke or transient ischemic attack.) The decision to bridge or not was left to the individual investigator. As a result, the literature offers diverse opinions about the appropriateness of transitioning to an alternative anticoagulant.41–43
Bridging does not make sense in most instances, since anticoagulants such as low-molecular-weight heparin have pharmacokinetics similar to those of the available TSOACs and also depend on renal clearance.41 However, there may be situations in which patients must be switched to a parenteral anticoagulant such as unfractionated or low-molecular-weight heparin. For example, if a TSOAC has to be held, the patient has acute renal failure, and a needed procedure is still several days away, it would be reasonable to start a heparin drip for an inpatient at increased thrombotic risk.
In patients with normal renal function, these alternative anticoagulants should be started at the time the next TSOAC dose would have been due.3,14,23 In patients with reduced renal function, initiation of an alternative anticoagulant may need to be delayed 12 to 48 hours depending on which TSOAC is being used, as well as on the degree of renal dysfunction. This delay would help ensure that the onset of anticoagulation with the alternative anticoagulant is timed with the offset of therapeutic anticoagulation with the TSOAC.
Although limited, information from available coagulation assays may assist with the timing of initiation of an alternative anticoagulant (see the following section on laboratory monitoring). Serial testing with appropriate coagulation assays may help identify when most of a TSOAC has been cleared from a patient.
Laboratory monitoring
Inevitably, some patients on TSOACs require urgent or emergency surgery. In certain situations, such as before an orthopedic spine procedure, in which the complications of bleeding could be devastating, it may be necessary to know if any residual anticoagulant effect is present.
Monitoring dabigatran. As one might expect, direct thrombin inhibitors such as dabigatran can prolong the prothrombin time and activated partial thromboplastin time (aPTT).44–47 However, the prothrombin time is not recommended for assessing the level of anticoagulation from dabigatran. Many institutions may be using a normal aPTT to rule out therapeutic concentrations of dabigatran, based on results from early in vitro and ex vivo studies.46 While appealing from a practical standpoint, practitioners should exercise caution when relying on the aPTT to assess the risk of perioperative bleeding. A more recent investigation in patients treated with dabigatran found that up to 35% of patients with a normal aPTT still had a plasma concentration in the therapeutic range.48
The thrombin time and ecarin clotting time are more sensitive tests for dabigatran. A normal thrombin time or ecarin clotting time indicates that no or only minimal dabigatran is present.48 Unfortunately, these two tests often are either unavailable or are associated with long turnaround times, which limits their usefulness in the perioperative setting.
Monitoring rivaroxaban and apixaban. Factor Xa inhibitors such as rivaroxaban and apixaban can also influence the prothrombin time and aPTT (Figure 1).44–47,49,50 The aPTT is relatively insensitive to these drugs at low concentrations. It has been suggested that a normal prothrombin time can reasonably exclude therapeutic concentrations of rivaroxaban.45,46 However, the effects on the prothrombin time are highly variable, changing with the reagent used.49,50 In addition, apixaban appears to have less impact on the prothrombin time overall. The INR is not recommended for monitoring the effect of factor Xa inhibitors.
Anti-factor Xa assays likely represent the best option to provide true quantitative information on the level of anticoagulation with either rivaroxaban or apixaban. However, the assays must be specifically calibrated for each drug for results to be useful. (Anti-factor Xa assays cannot be used for heparin or low-molecular-weight heparin.) Further, most institutions do not yet have this capability. When appropriately calibrated, normal anti-factor Xa levels would exclude any effect of rivaroxaban or apixaban.
Reversal of anticoagulation
If patients on TSOACs require emergency surgery or present with significant bleeding in the setting of persistent anticoagulation, it may be necessary to try to reverse the anticoagulation.
Unlike warfarin or heparin, TSOACS do not have specific reversal agents, though specific antidotes are being developed. For example, researchers are evaluating antibodies capable of neutralizing dabigatran, as well as recombinant thrombin and factor Xa molecules that could antagonize dabigatran and rivaroxaban, respectively.51–53
Reversal can be attempted by neutralizing or removing the offending drug. Activated charcoal may be able to reduce absorption of TSOACs that were recently ingested,44 and dabigatran can be removed by hemodialysis.
However, certain practical considerations may limit the use of dialysis in the perioperative period. Insertion of a temporary dialysis line in an anticoagulated patient poses additional bleeding risks. A standard 4-hour hemodialysis session may remove only about 70% of dabigatran from the plasma, which may not be enough to prevent perioperative bleeding.54 Dabigatran also tends to redistribute from adipose tissue back into plasma after each dialysis session.55 Serial sessions of high-flux intermittent hemodialysis or continuous renal replacement therapy may therefore be needed to counteract rebound elevations in the dabigatran concentration.
Reversal can also be attempted through activation of the coagulation cascade via other mechanisms. Fresh-frozen plasma is unlikely to be a practical solution for reversal.44 Although it can readily replace the clotting factors depleted by vitamin K antagonists, large volumes of fresh-frozen plasma would be needed to overwhelm thrombin or factor Xa inhibition by TSOACs.
There are limited data on the use of prothrombin complex concentrates or recombinant activated factor VIIa in patients on TSOACs, though their use can be considered.56 In a trial in 12 healthy participants, a nonactivated four-factor prothrombin complex concentrate containing factors II, VII, IX, and X immediately and completely reversed the anticoagulant effect of rivaroxaban but had no effect on dabigatran.57 Before 2013, there were no nonactivated four-factor prothrombin complex concentrates available in the United States. The FDA has since approved Kcentra for the urgent reversal of vitamin K antagonists, meaning that the reversal of TSOACs in major bleeding events would still be off-label.58 Giving any of the clotting factors carries a risk of thromboembolism.
Resumption of anticoagulation
TSOACs have a rapid onset of action, and therapeutic levels are reached within a few hours of administration.
Extrapolating from the ACCP guidelines, TSOACs can generally be restarted at therapeutic doses 24 hours after low-bleeding-risk procedures.2 Therapeutic dosing should be delayed 48 to 72 hours after a procedure with a high bleeding risk, assuming adequate hemostasis has been achieved. Prophylactic unfractionated heparin or low-molecular-weight heparin therapy can be given in the interim if deemed safe. Alternatively, for orthopedic patients ultimately transitioning back to therapeutic rivaroxaban after hip or knee arthroplasty, prophylactic rivaroxaban doses can be started 6 to 10 hours after surgery.14
There are numerous reasons why the resumption of TSOACs may have to be delayed after surgery, including nothing-by-mouth status, postoperative nausea and vomiting, ileus, gastric or bowel resection, and the anticipated need for future procedures. Since dabigatran capsules cannot be crushed, they cannot be given via nasogastric tube in patients with postoperative dysphagia. Parenteral anticoagulants should be used until these issues resolve.
Unfractionated heparin is still the preferred anticoagulant in unstable or potentially unstable patients, given its ease of monitoring, quick offset of action, and reversibility. When patients have stabilized, TSOACs can be resumed when the next dose of low-molecular-weight heparin would have been due or when the unfractionated heparin drip is discontinued.3,14,23
UNTIL EVIDENCE-BASED GUIDELINES ARE DEVELOPED
The development of TSOACs has ushered in an exciting new era for anticoagulant therapy. Providers involved in perioperative medicine will increasingly encounter patients on dabigatran, rivaroxaban, and apixaban. However, until evidence-based guidelines are developed for these new anticoagulants, clinicians will have to apply their knowledge of pharmacology and critically evaluate expert recommendations in order to manage patients safely throughout the perioperative period.
More then 2.5 million patients in the United States are on long-term anticoagulation therapy for atrial fibrillation, venous thromboembolic disease, or mechanical heart valves,1 and the number is expected to rise as the population ages. Each year, about 10% of these patients undergo an invasive procedure or surgery that requires temporary interruption of anticoagulation.2
Most physicians are familiar with the perioperative management of warfarin, a vitamin K antagonist, since for decades it has been the sole oral anticoagulant available. However, many physicians lack experience with the three target-specific oral anticoagulants (TSOACs; also known as “novel” oral anticoagulants) approved so far: the direct thrombin inhibitor dabigatran (Pradaxa) and the direct factor Xa inhibitors rivaroxaban (Xarelto) and apixaban (Eliquis).
With their rapid onset of action, predictable pharmacokinetics, relatively short half-lives, and fewer drug-drug interactions than warfarin, TSOACs overcome many of the limitations of the older oral anticoagulant warfarin. In many ways, these qualities simplify the perioperative management of anticoagulation. At the same time, these new drugs also bring new challenges: caution is needed in patients with renal impairment; the level of anticoagulation is difficult to assess; and there is no specific antidote or standardized procedure to reverse their anticoagulant effect. While various periprocedural protocols for TSOAC therapy have been proposed, evidence-based guidelines are still to come.
This article first discusses the pharmacology of dabigatran, rivaroxaban, and apixaban that is pertinent to the perioperative period. It then briefly reviews the general principles of perioperative management of anticoagulation. The final section provides specific recommendations for the perioperative management of TSOACs.
PHARMACOLOGY OF TARGET-SPECIFIC ORAL ANTICOAGULANTS
Dabigatran, a factor IIa inhibitor
Dabigatran is an oral direct thrombin (factor IIa) inhibitor. It exerts its anticoagulant effect by blocking the generation of fibrin, inhibiting platelet aggregation, and dampening the activity of factors V, VIII, and XI (Figure 1).3,4 From its introduction in October 2010 through August 2012, nearly 3.7 million prescriptions were dispensed to 725,000 patients in the United States.5
Indications for dabigatran. Dabigatran is approved in the United States and Canada for preventing stroke in nonvalvular atrial fibrillation (Table 1).6 More recently, it received US approval for treating deep vein thrombosis or pulmonary embolism after 5 to 10 days of a parenteral anticoagulant.7,8 It is also approved in Europe and Canada for preventing venous thromboembolism (VTE) after total hip replacement and knee arthroplasty.9,10
Dabigatran is contraindicated in patients with a mechanical heart valve, based on a phase 2 study in which it conferred a higher risk of thromboembolism and bleeding than warfarin.3,11
Pharmacokinetics of dabigatran. Dabigatran is formulated as a prodrug, dabigatran etexilate, in a capsule containing multiple small pellets.12 The capsules should not be crushed, as this significantly increases oral bioavailability. The prodrug is absorbed across the gastric mucosa and is then rapidly converted to the active form (Table 2).
Plasma concentrations peak within 2 hours of ingestion, which means that therapeutic anticoagulation is achieved shortly after taking the drug.
Only 35% of dabigatran is protein-bound, which allows it to be removed by hemodialysis. Nearly 85% of the drug is eliminated in the urine. It has a half-life of 13 to 15 hours in patients with normal renal function.3 However, its half-life increases to about 27 hours in patients whose creatinine clearance is less than 30 mL/min. As a result, the dose must be reduced in patients with renal impairment (Table 1).
Dabigatran is not metabolized by the cytochrome P450 enzymes, but it is a substrate for P-glycoprotein, so it still has the potential for drug-drug interactions.3 Practitioners should be familiar with these potential interactions (Table 3), as they can result in higher- or lower-than-expected plasma concentrations of dabigatran in the perioperative period.13
Rivaroxaban, a factor Xa inhibitor
Rivaroxaban is an oral direct factor Xa inhibitor. It has been approved by the US Food and Drug Administration (FDA) for the prevention of stroke in nonvalvular atrial fibrillation, for VTE treatment, and for VTE prophylaxis after hip replacement or knee replacement (Table 1).14–20 It has not yet been studied in patients with hip fracture.
Pharmacokinetics of rivaroxaban. Rivaroxaban is manufactured as a tablet that is best absorbed in the stomach (Table 2).14 In contrast to dabigatran, it can be crushed and, for example, mixed with applesauce for patients who have trouble swallowing. It can also be mixed with water and given via nasogastric tube; however, postpyloric administration should be avoided.
Plasma concentrations peak within a few hours after ingestion. Rivaroxaban is highly protein-bound, so it cannot be eliminated by hemodialysis.
The drug relies on renal elimination to a smaller degree than dabigatran, with one-third of the dose eliminated unchanged in the urine, one-third eliminated in the urine as inactive metabolite, and the remaining one-third eliminated in the feces. However, enough parent compound is cleared through the kidneys that the half-life of rivaroxaban increases from 8.3 hours in healthy individuals to 9.5 hours in patients whose creatinine clearance is less than 30 mL/min.21 As with dabigatran, the dose must be adjusted for renal impairment (Table 1).
Rivaroxaban has significant liver metabolism, specifically through the cytochrome P450 3A4 enzyme, and it is also a substrate of P-glycoprotein. Therefore, potential drug-drug interactions must be taken into account, as they may lead to important alterations in plasma concentrations (Table 3).
Apixaban, a factor Xa inhibitor
Apixaban is also an oral direct factor Xa inhibitor. It is the newest of the oral anticoagulants to be approved in the United States, specifically for preventing stroke in nonvalvular atrial fibrillation (Table 1).22
Pharmacokinetics of apixaban. Apixaban is produced as a tablet that is absorbed slowly through the gastrointestinal tract, mainly the distal small bowel and ascending colon (Table 2).23
Peak plasma concentrations are reached a few hours after ingestion. Like rivaroxaban, apixaban is highly protein-bound, so it cannot be removed by hemodialysis.
Apixaban is similar to rivaroxaban in that 27% of the parent compound is cleared through the kidneys, it undergoes significant hepatic metabolism through cytochrome P450 3A4, and it is a substrate for P-glycoprotein.
Drug-drug interactions must be considered as a potential source of altered drug exposure and clearance (Table 3).
Unlike dabigatran and rivaroxaban, dose reduction is not based on the calculated creatinine clearance. Instead, a reduced dose is required if the patient meets two of the following three criteria:
- Serum creatinine level ≥ 1.5 mg/dL
- Age ≥ 80
- Weight ≤ 60 kg (Table 1).
The American Heart Association/American Stroke Association guidelines further recommend against using apixaban in patients with a creatinine clearance less than 25 mL/min.24
Edoxaban, a factor Xa inhibitor in development
Edoxaban (Savaysa), another factor Xa inhibitor, is available in Japan and has been submitted for approval in the United States for treating VTE and for preventing stroke in patients with
PERIOPERATIVE CONSIDERATIONS IN ANTICOAGULATION
Before addressing the perioperative management of TSOACs, let us review the evidence guiding the perioperative management of any chronic anticoagulant.
In fact, no large prospective randomized trial has clearly defined the risks and benefits of using or withholding a bridging anticoagulation strategy around surgery and other procedures, though the PERIOP 2 and BRIDGE trials are currently ongoing.25,26 There are some data regarding continuing anticoagulation without interruption, but they have mainly been derived from specific groups (eg, patients on warfarin undergoing cardiac pacemaker or defibrillator placement) and in procedures that pose a very low risk of bleeding complications (eg, minor dental extractions, cataract surgery, dermatologic procedures).2,27 Recommendations are, therefore, necessarily based on small perioperative trials and data gleaned from cohort review and from studies that did not involve surgical patients.
Ultimately, the decisions whether to discontinue oral anticoagulants and whether to employ bridging anticoagulation are based on assumptions about the risks of bleeding and the risk of thrombotic events, with similar assumptions regarding the effects of anticoagulants on both outcomes. In addition, the relative acceptance of bleeding vs thrombotic risks implicitly guides these complex decisions.
Perioperative bleeding risk
Many risk factors specific to the patient and to the type of surgery affect the rates and severity of perioperative bleeding.28
As for patient-specific risk factors, a small retrospective cohort analysis revealed that a HAS-BLED score of 3 or higher was highly discriminating in predicting perioperative bleeding in atrial fibrillation patients receiving anticoagulation.29 (The HAS-BLED score is based on hypertension, abnormal renal or liver function, stroke, bleeding, labile international normalized ratio [INR], elderly [age > 65] and drug therapy.30) However, there are no widely validated tools that incorporate patient-specific factors to accurately predict bleeding risk in an individual patient.
Therefore, the American College of Chest Physicians (ACCP) guidelines suggest coarsely categorizing bleeding risk as either low or high solely on the basis of the type of procedure.2 Procedures considered “high-risk” have a risk greater than 1.5% to 2% and include urologic surgery involving the prostate or kidney, colonic polyp resections, surgeries involving highly vascular organs such as the liver or spleen, joint replacements, cancer surgeries, and cardiac or neurosurgical procedures.
Perioperative thrombotic risk
The ACCP guidelines2 place patients with atrial fibrillation, VTE, or mechanical heart valves in three risk groups for perioperative thromboembolism without anticoagulation, based on their annual risk of a thrombotic event:
- High risk—annual risk of a thrombotic event > 10%
- Moderate risk—5% to 10%
- Low risk—< 5%.
Comparing the risks calculated by these methods with the real-world risk of perioperative thrombosis highlights the problem of applying nonperioperative risk calculations: the perioperative period exposes patients to a higher risk than these models would predict.31 Nonetheless, these risk categorizations likely have some validity in stratifying patients into risk groups, even if the absolute risks are inaccurate.
Perioperative bridging for patients taking warfarin
Many patients with atrial fibrillation, VTE, or a mechanical heart valve need to interrupt their warfarin therapy because of the bleeding risk of an upcoming procedure.
The perioperative management of warfarin and other vitamin K antagonists is challenging because of the pharmacokinetics and pharmacodynamics of these drugs. Because it has a long half-life, warfarin usually must be stopped 4 to 5 days before a procedure in order to allow not only adequate clearance of the drug itself, but also restoration of functional clotting factors to normal or near-normal levels.12 Warfarin can generally be resumed 12 to 24 hours after surgery, assuming adequate hemostasis has been achieved, and it will again take several days for the INR to reach the therapeutic range.
The ACCP guidelines recommend using the perioperative risk of thromboembolism to make decisions about the need for bridging anticoagulation during warfarin interruption.2 They suggest that patients at high risk of thrombosis receive bridging with an alternative anticoagulant such as low-molecular-weight heparin or unfractionated heparin, because of the prolonged duration of subtherapeutic anticoagulation.
There has been clinical interest in using a TSOAC instead of low-molecular-weight or unfractionated heparin for bridging in the perioperative setting. Although this approach may be attractive from a cost and convenience perspective, it cannot be endorsed as yet because of the lack of information on the pros and cons of such an approach.
Patients at low thrombotic risk do not require bridging. In patients at moderate risk, the decision to bridge or not to bridge is based on careful consideration of patient-specific and surgery-specific factors.
PERIOPERATIVE MANAGEMENT OF TARGET-SPECIFIC ORAL ANTICOAGULANTS
As summarized above, the perioperative management strategy for chronic anticoagulation is based on limited evidence, even for drugs as well established as warfarin.
The most recent ACCP guidelines on the perioperative management of antithrombotic therapy do not mention TSOACs.2 For now, the management strategy must be based on the pharmacokinetics of the drugs, package inserts from the manufacturers, and expert recommendations.3,14,23,32–34 Fortunately, because TSOACs have a more favorable pharmacokinetic profile than that of warfarin, their perioperative uses should be more streamlined. As always, the goal is to minimize the risk of both periprocedural bleeding and thromboembolism.
Timing of cessation of anticoagulation
The timing of cessation of TSOACs before an elective procedure depends primarily on two factors: the bleeding risk of the procedure and the patient’s renal function. Complete clearance of the medication is not necessary in all circumstances.
TSOACs should be stopped four to five half-lives before a procedure with a high bleeding risk, so that there is no or only minimal residual anticoagulant effect. The drug can be stopped two to three half-lives before a procedure with a low bleeding risk. Remember: the half-life increases as creatinine clearance decreases.
Specific recommendations may vary across institutions, but a suggested strategy is shown in Table 4.3,4,21,23,32–35 For the small subset of patients on P-glycoprotein or cytochrome P450 inhibitors or inducers, further adjustment in the time of discontinuation may be required.
Therapy does not need to be interrupted for procedures with a very low bleeding risk, as defined above.33,34 There is also preliminary evidence that TSOACs, similar to warfarin, may be continued during cardiac pacemaker or defibrillator placement.36
Evidence from clinical trials of perioperative TSOAC management
While the above recommendations are logical, studies are needed to prospectively evaluate perioperative management strategies.
The RE-LY trial (Randomized Evaluation of Long-Term Anticoagulation Therapy), which compared the effects of dabigatran and warfarin in preventing stroke in patients with atrial fibrillation, is one of the few clinical trials that also looked at periprocedural bleeding.37 About a quarter of the RE-LY participants required interruption of anticoagulation for a procedure.
Warfarin was managed according to local practices. For most of the study, the protocol required that dabigatran be discontinued 24 hours before a procedure, regardless of renal function or procedure type. The protocol was later amended and closely mirrored the management plan outlined in Table 4.
With either protocol, there was no statistically significant difference between dabigatran and warfarin in the rates of bleeding and thrombotic complications in the 7 days before or 30 days after the procedure.
A major limitation of the study was that most patients underwent a procedure with a low bleeding risk, so the analysis was likely underpowered to evaluate rates of bleeding in higher-risk procedures.
The ROCKET-AF trial (Rivaroxaban Once-daily Oral Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation) also shed light on periprocedural bleeding.15 About 15% of the participants required temporary interruption of anticoagulation for a surgical or invasive procedure.38
The study protocol called for discontinuing rivaroxaban 2 days before any procedure. Warfarin was to be held for 4 days to achieve a goal INR of 1.5 or less.15
Rates of major and nonmajor clinically significant bleeding at 30 days were similar with rivaroxaban and with warfarin.38 As with the RE-LY trial, the retrospective analysis was probably underpowered for assessing rates of bleeding in procedures with higher risk.
Perioperative bridging
While stopping a TSOAC in the perioperative period decreases the risk of bleeding, it naturally increases the risk of thromboembolism. However, patients on TSOACs should not routinely require perioperative bridging with an alternative anticoagulant, regardless of thrombotic risk.
Of note, dabigatran, rivaroxaban, and apixaban carry black-box warnings that discontinuation places patients at higher risk of thrombotic events.3,14,23 These warnings further state that coverage with an alternative anticoagulant should be strongly considered during interruption of therapy for reasons other than pathologic bleeding.
However, it does not necessarily follow that perioperative bridging is required. For example, the warning for rivaroxaban is based on the finding in the ROCKET-AF trial that patients in the rivaroxaban group had higher rates of stroke than those in the warfarin group after the study drugs were stopped at the end of the trial.39 While there was initial concern that this could represent a prothrombotic rebound effect, the authors subsequently showed that patients in the rivaroxaban group were more likely to have had a subtherapeutic INR when transitioning to open-label vitamin-K-antagonist therapy.39,40 There was no difference in the rate of stroke or systemic embolism between the rivaroxaban and warfarin groups when anticoagulation was temporarily interrupted for a procedure.38
The risks and benefits of perioperative bridging with TSOACs are difficult to evaluate, given the dearth of trial data. In the RE-LY trial, only 17% of patients on dabigatran and 28% of patients on warfarin underwent periprocedural bridging.37 The selection criteria and protocol for bridging were not reported. In the ROCKET-AF trial, only 9% of patients received bridging therapy despite a mean CHADS2 score of 3.4.38 (The CHADS2 score is calculated as 1 point each for congestive heart failure, hypertension, age ≥ 75, and diabetes; 2 points for stroke or transient ischemic attack.) The decision to bridge or not was left to the individual investigator. As a result, the literature offers diverse opinions about the appropriateness of transitioning to an alternative anticoagulant.41–43
Bridging does not make sense in most instances, since anticoagulants such as low-molecular-weight heparin have pharmacokinetics similar to those of the available TSOACs and also depend on renal clearance.41 However, there may be situations in which patients must be switched to a parenteral anticoagulant such as unfractionated or low-molecular-weight heparin. For example, if a TSOAC has to be held, the patient has acute renal failure, and a needed procedure is still several days away, it would be reasonable to start a heparin drip for an inpatient at increased thrombotic risk.
In patients with normal renal function, these alternative anticoagulants should be started at the time the next TSOAC dose would have been due.3,14,23 In patients with reduced renal function, initiation of an alternative anticoagulant may need to be delayed 12 to 48 hours depending on which TSOAC is being used, as well as on the degree of renal dysfunction. This delay would help ensure that the onset of anticoagulation with the alternative anticoagulant is timed with the offset of therapeutic anticoagulation with the TSOAC.
Although limited, information from available coagulation assays may assist with the timing of initiation of an alternative anticoagulant (see the following section on laboratory monitoring). Serial testing with appropriate coagulation assays may help identify when most of a TSOAC has been cleared from a patient.
Laboratory monitoring
Inevitably, some patients on TSOACs require urgent or emergency surgery. In certain situations, such as before an orthopedic spine procedure, in which the complications of bleeding could be devastating, it may be necessary to know if any residual anticoagulant effect is present.
Monitoring dabigatran. As one might expect, direct thrombin inhibitors such as dabigatran can prolong the prothrombin time and activated partial thromboplastin time (aPTT).44–47 However, the prothrombin time is not recommended for assessing the level of anticoagulation from dabigatran. Many institutions may be using a normal aPTT to rule out therapeutic concentrations of dabigatran, based on results from early in vitro and ex vivo studies.46 While appealing from a practical standpoint, practitioners should exercise caution when relying on the aPTT to assess the risk of perioperative bleeding. A more recent investigation in patients treated with dabigatran found that up to 35% of patients with a normal aPTT still had a plasma concentration in the therapeutic range.48
The thrombin time and ecarin clotting time are more sensitive tests for dabigatran. A normal thrombin time or ecarin clotting time indicates that no or only minimal dabigatran is present.48 Unfortunately, these two tests often are either unavailable or are associated with long turnaround times, which limits their usefulness in the perioperative setting.
Monitoring rivaroxaban and apixaban. Factor Xa inhibitors such as rivaroxaban and apixaban can also influence the prothrombin time and aPTT (Figure 1).44–47,49,50 The aPTT is relatively insensitive to these drugs at low concentrations. It has been suggested that a normal prothrombin time can reasonably exclude therapeutic concentrations of rivaroxaban.45,46 However, the effects on the prothrombin time are highly variable, changing with the reagent used.49,50 In addition, apixaban appears to have less impact on the prothrombin time overall. The INR is not recommended for monitoring the effect of factor Xa inhibitors.
Anti-factor Xa assays likely represent the best option to provide true quantitative information on the level of anticoagulation with either rivaroxaban or apixaban. However, the assays must be specifically calibrated for each drug for results to be useful. (Anti-factor Xa assays cannot be used for heparin or low-molecular-weight heparin.) Further, most institutions do not yet have this capability. When appropriately calibrated, normal anti-factor Xa levels would exclude any effect of rivaroxaban or apixaban.
Reversal of anticoagulation
If patients on TSOACs require emergency surgery or present with significant bleeding in the setting of persistent anticoagulation, it may be necessary to try to reverse the anticoagulation.
Unlike warfarin or heparin, TSOACS do not have specific reversal agents, though specific antidotes are being developed. For example, researchers are evaluating antibodies capable of neutralizing dabigatran, as well as recombinant thrombin and factor Xa molecules that could antagonize dabigatran and rivaroxaban, respectively.51–53
Reversal can be attempted by neutralizing or removing the offending drug. Activated charcoal may be able to reduce absorption of TSOACs that were recently ingested,44 and dabigatran can be removed by hemodialysis.
However, certain practical considerations may limit the use of dialysis in the perioperative period. Insertion of a temporary dialysis line in an anticoagulated patient poses additional bleeding risks. A standard 4-hour hemodialysis session may remove only about 70% of dabigatran from the plasma, which may not be enough to prevent perioperative bleeding.54 Dabigatran also tends to redistribute from adipose tissue back into plasma after each dialysis session.55 Serial sessions of high-flux intermittent hemodialysis or continuous renal replacement therapy may therefore be needed to counteract rebound elevations in the dabigatran concentration.
Reversal can also be attempted through activation of the coagulation cascade via other mechanisms. Fresh-frozen plasma is unlikely to be a practical solution for reversal.44 Although it can readily replace the clotting factors depleted by vitamin K antagonists, large volumes of fresh-frozen plasma would be needed to overwhelm thrombin or factor Xa inhibition by TSOACs.
There are limited data on the use of prothrombin complex concentrates or recombinant activated factor VIIa in patients on TSOACs, though their use can be considered.56 In a trial in 12 healthy participants, a nonactivated four-factor prothrombin complex concentrate containing factors II, VII, IX, and X immediately and completely reversed the anticoagulant effect of rivaroxaban but had no effect on dabigatran.57 Before 2013, there were no nonactivated four-factor prothrombin complex concentrates available in the United States. The FDA has since approved Kcentra for the urgent reversal of vitamin K antagonists, meaning that the reversal of TSOACs in major bleeding events would still be off-label.58 Giving any of the clotting factors carries a risk of thromboembolism.
Resumption of anticoagulation
TSOACs have a rapid onset of action, and therapeutic levels are reached within a few hours of administration.
Extrapolating from the ACCP guidelines, TSOACs can generally be restarted at therapeutic doses 24 hours after low-bleeding-risk procedures.2 Therapeutic dosing should be delayed 48 to 72 hours after a procedure with a high bleeding risk, assuming adequate hemostasis has been achieved. Prophylactic unfractionated heparin or low-molecular-weight heparin therapy can be given in the interim if deemed safe. Alternatively, for orthopedic patients ultimately transitioning back to therapeutic rivaroxaban after hip or knee arthroplasty, prophylactic rivaroxaban doses can be started 6 to 10 hours after surgery.14
There are numerous reasons why the resumption of TSOACs may have to be delayed after surgery, including nothing-by-mouth status, postoperative nausea and vomiting, ileus, gastric or bowel resection, and the anticipated need for future procedures. Since dabigatran capsules cannot be crushed, they cannot be given via nasogastric tube in patients with postoperative dysphagia. Parenteral anticoagulants should be used until these issues resolve.
Unfractionated heparin is still the preferred anticoagulant in unstable or potentially unstable patients, given its ease of monitoring, quick offset of action, and reversibility. When patients have stabilized, TSOACs can be resumed when the next dose of low-molecular-weight heparin would have been due or when the unfractionated heparin drip is discontinued.3,14,23
UNTIL EVIDENCE-BASED GUIDELINES ARE DEVELOPED
The development of TSOACs has ushered in an exciting new era for anticoagulant therapy. Providers involved in perioperative medicine will increasingly encounter patients on dabigatran, rivaroxaban, and apixaban. However, until evidence-based guidelines are developed for these new anticoagulants, clinicians will have to apply their knowledge of pharmacology and critically evaluate expert recommendations in order to manage patients safely throughout the perioperative period.
- Douketis JD, Berger PB, Dunn AS, et al; American College of Chest Physicians. The perioperative management of antithrombotic therapy: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):299S–339S.
- Douketis JD, Spyropoulos AC, Spencer FA, et al; American College of Chest Physicians. Perioperative management of antithrombotic therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e326S–e350S.
- Boehringer Ingelheim Pharmaceuticals, Inc. PRADAXA (dabigatran) package insert. http://bidocs.boehringer-ingelheim.com/BIWebAc-cess/ViewServlet.ser?docBase=renetnt&folderPath=/Prescribing%20Information/PIs/Pradaxa/Pradaxa.pdf. Accessed August 6, 2014.
- Levy JH, Faraoni D, Spring JL, Douketis JD, Samama CM. Managing new oral anticoagulants in the perioperative and intensive care unit setting. Anesthesiology 2013; 118:1466–1474.
- US Food and Drug Administration (FDA). FDA drug safety communication: update on the risk for serious bleeding events with the anticoagulant Pradaxa (dabigatran). www.fda.gov/drugs/drugsafety/ucm326580.htm. Accessed August 6, 2014.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- Schulman S, Kearon C, Kakkar AK, et al; RE-MEDY Trial Investigators. Extended use of dabigatran, warfarin, or placebo in venous thromboembolism. N Engl J Med 2013; 368:709–718.
- Eriksson BI, Dahl OE, Rosencher N, Büller HR, et al; RE-NOVATE Study Group. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement: a randomised, double-blind, non-inferiority trial. Lancet 2007; 370:949–956.
- Eriksson BI, Dahl OE, Rosencher N, et al; RE-MODEL Study Group. Oral dabigatran etexilate vs subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: the RE-MODEL randomized trial. J Thromb Haemost 2007; 5:2178–2185.
- Eikelboom JW, Connolly SJ, Brueckmann M, et al; RE-ALIGN Investigators. Dabigatran versus warfarin in patients with mechanical heart valves. N Engl J Med 2013; 369:1206–1214.
- Ageno W, Gallus AS, Wittkowsky A, Crowther M, Hylek EM, Palareti G; American College of Chest Physicians. Oral anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e44S–e88S.
- Blech S, Ebner T, Ludwig-Schwellinger E, Stangier J, Roth W. The metabolism and disposition of the oral direct thrombin inhibitor, dabigatran, in humans. Drug Metab Dispos 2008; 36:386–399.
- Janssen Pharmaceuticals, Inc. XARELTO (rivaroxaban) package insert. www.xareltohcp.com/about-xarelto/about-xarelto.html?utm_source=google&utm_medium=cpc&utm_campaign=Branded+-+Broad&utm_term=xarelto%20rivaroxaban&utm_content=Xarelto+Rivaroxaban|mkwid|sxSDxPb4m_dc|pcrid|34667840494. Accessed August 6, 2014.
- Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–391.
- EINSTEIN Investigators; Bauersachs R, Berkowitz SD, Brenner B, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med 2010; 363:2499–2510.
- EINSTEIN–PE Investigators; Büller HR, Prins MH, Lensin AW, et al. Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N Engl J Med 2012; 366:1287–1297.
- Eriksson BI, Borris LC, Friedman RJ, et al; RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med 2008; 358:2765–2775.
- Kakkar AK, Brenner B, Dahl OE, et al; RECORD2 Investigators. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet 2008; 372:31–39.
- Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med 2008; 358:2776–2786.
- Kubitza D, Becka M, Mueck W, et al. Effects of renal impairment on the pharmacokinetics, pharmacodynamics and safety of rivaroxaban, an oral, direct factor Xa inhibitor. Br J Clin Pharmacol 2010; 70:703–712.
- Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365:981–992.
- Bristol-Myers Squibb Company. ELIQUIS (apixaban) package insert. www.eliquis.com/index.aspx. Accessed August 6, 2014.
- Furie KL, Goldstein LB, Albers GW, et al; American Heart Association Stroke Council. Oral antithrombotic agents for the prevention of stroke in nonvalvular atrial fibrillation: a science advisory for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:3442–3453.
- ClinicalTrials.gov, US National Institutes of Health. PERIOP 2 - A Safety and Effectiveness Study of LMWH Bridging Therapy Versus Placebo Bridging Therapy for Patients on Long Term Warfarin and Require Temporary Interruption of Their Warfarin. http://clinicaltri-als.gov/show/NCT00432796. Accessed August 6, 2014.
- ClinicalTrials.gov, US National Institutes of Health. Effectiveness of Bridging Anticoagulation for Surgery (The BRIDGE Study). http://clinicaltrials.gov/ct2/show/NCT00786474. Accessed August 6, 2014.
- Birnie DH, Healey JS, Wells GA, et al; BRUISE CONTROL Investigators. Pacemaker or defibrillator surgery without interruption of anticoagulation. N Engl J Med 2013; 368:2084–2093.
- Oberweis BS, Nukala S, Rosenberg A, et al. Thrombotic and bleeding complications after orthopedic surgery. Am Heart J 2013; 165:427.e1–433.e1.
- Omran H, Bauersachs R, Rübenacker S, Goss F, Hammerstingl C. The HAS-BLED score predicts bleedings during bridging of chronic oral anticoagulation. Results from the national multicentre BNK Online bRiDging REgistRy (BORDER). Thromb Haemost 2012; 108:65–73.
- Pisters R, Lane DA, Nieuwlaaat R, de Vos CB, Crijns HJGM, Lip GYH. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation. The Euro Heart Survey. Chest 2010; 138:1093–1100.
- Kaatz S, Douketis JD, Zhou H, Gage BF, White RH. Risk of stroke after surgery in patients with and without chronic atrial fibrillation. J Thromb Haemost 2010; 8:884–890.
- van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost 2010; 103:1116–1127.
- Connolly G, Spyropoulos AC. Practical issues, limitations, and periprocedural management of the NOAC’s. J Thromb Thrombolysis 2013; 36:212–222.
- Spyropoulos AC, Douketis JD. How I treat anticoagulated patients undergoing an elective procedure or surgery. Blood 2012; 120:2954–2962.
- Kaatz S, Kouides PA, Garcia DA, et al. Guidance on the emergent reversal of oral thrombin and factor Xa inhibitors. Am J Hematol 2012; 87(suppl 1):S141–S145.
- Rowley CP, Bernard ML, Brabham WW, et al. Safety of continuous anticoagulation with dabigatran during implantation of cardiac rhythm devices. Am J Cardiol 2013; 111:1165–1168.
- Healey JS, Eikelboom J, Douketis J, et al; RE-LY Investigators. Periprocedural bleeding and thromboembolic events with dabigatran compared with warfarin: results from the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) randomized trial. Circulation 2012; 126:343–348.
- Sherwood MW, Douketis JD, Patel MR, et al; on behalf of the ROCKET AF Investigators. Outcomes of temporary interruption of rivaroxaban compared with warfarin in patients with nonvalvular atrial fibrillation: results from the Rivaroxaban Once Daily, Oral, Direct Factor Xa Inhibition Compared with Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation (ROCKET AF). Circulation 2014; 129:1850–1859.
- Patel MR, Hellkamp AS, Lokhnygina Y, et al. Outcomes of discontinuing rivaroxaban compared with warfarin in patients with nonvalvular atrial fibrillation: analysis from the ROCKET AF trial (Rivaroxaban Once-Daily, Oral, Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation). J Am Coll Cardiol 2013; 61:651–658.
- Reynolds MR. Discontinuation of rivaroxaban: filling in the gaps. J Am Coll Cardiol 2013; 61:659–660.
- Turpie AG, Kreutz R, Llau J, Norrving B, Haas S. Management consensus guidance for the use of rivaroxaban—an oral, direct factor Xa inhibitor. Thromb Haemost 2012; 108:876–886.
- Gallego P, Apostolakis S, Lip GY. Bridging evidence-based practice and practice-based evidence in periprocedural anticoagulation. Circulation 2012; 126:1573–1576.
- Sié P, Samama CM, Godier A, et al; Working Group on Perioperative Haemostasis. Surgery and invasive procedures in patients on long-term treatment with direct oral anticoagulants: thrombin or factor-Xa inhibitors. Recommendations of the Working Group on Perioperative Haemostasis and the French Study Group on Thrombosis and Haemostasis. Arch Cardiovasc Dis 2011; 104:669–676.
- King CS, Holley AB, Moores LK. Moving toward a more ideal anticoagulant: the oral direct thrombin and factor Xa inhibitors. Chest 2013; 143:1106–1116.
- Baglin T, Hillarp A, Tripodi A, Elalamy I, Buller H, Ageno W. Measuring oral direct inhibitors (ODIs) of thrombin and factor Xa: a recommendation from the Subcommittee on Control of Anticoagulation of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2013; 11:756–760.
- Baglin T, Keeling D, Kitchen S; British Committee for Standards in Haematology. Effects on routine coagulation screens and assessment of anticoagulant intensity in patients taking oral dabigatran or rivaroxaban: guidance from the British Committee for Standards in Haematology. Br J Haematol 2012; 159:427–429.
- Mani H, Kasper A, Lindhoff-Last E. Measuring the anticoagulant effects of target specific oral anticoagulants—reasons, methods and current limitations. J Thromb Thrombolysis 2013; 36:187–194.
- Hawes EM, Deal AM, Funk-Adcock D, et al. Performance of coagulation tests in patients on therapeutic doses of dabigatran: a cross-sectional pharmacodynamic study based on peak and trough plasma levels. J Thromb Haemost 2013; 11:1493–1502.
- Barrett YC, Wang Z, Frost C, Shenker A. Clinical laboratory measurement of direct factor Xa inhibitors: anti-Xa assay is preferable to prothrombin time assay. Thromb Haemost 2010; 104:1263–1271.
- Smythe MA, Fanikos J, Gulseth MP, et al. Rivaroxaban: practical considerations for ensuring safety and efficacy. Pharmacotherapy 2013; 33:1223–1245.
- Van Ryn J, Litzenburger T, Waterman A, et al. Dabigatran anticoagulant activity is neutralized by an antibody selective to dabigatran in in vitro and in vivo models. J Am Coll Cardiol 2011; 57:E1130.
- Sheffield W, Lambourne M, Bhakta V, Eltringham-Smith L, Arnold D, Crowther M. Active site-mutated thrombin S195A but not active site-blocked thrombin counteracts the anticoagulant activity of dabigatran in plasma. Abstract presented at the International Society of Thrombosis and Haemostasis 2013 Congress. http://onlinelibrary.wiley.com/doi/10.1111/jth.2013.11.issue-s2/issuetoc. Accessed August 6, 2014.
- Lu G, Luan P, Hollenbach SJ, et al. Reconstructed recombinant factor Xa as an antidote to reverse anticoagulation by factor Xa inhibitors (abstract). J Thromb Haemost 2009; 7(suppl 2):abstract OC-TH-107.
- Stangier J, Rathgen K, Stähle H, Mazur D. Influence of renal impairment on the pharmacokinetics and pharmacodynamics of oral dabigatran etexilate: an open-label, parallel-group, single-centre study. Clin Pharmacokinet 2010; 49:259–268.
- Singh T, Maw TT, Henry BL, et al. Extracorporeal therapy for dabigatran removal in the treatment of acute bleeding: a single center experience. Clin J Am Soc Nephrol 2013; 8:1533–1539.
- Kaatz S, Crowther M. Reversal of target-specific oral anticoagulants. J Thromb Thrombolysis 2013; 36:195–202.
- Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation 2011; 124:1573–1579.
- Sarode R, Milling TJ, Refaai MA, et al. Efficacy and safety of a 4-factor prothrombin complex concentrate in patients on vitamin K antagonists presenting with major bleeding: a randomized, plasma-controlled, phase IIIb study. Circulation 2013; 128:1234–1243.
- Douketis JD, Berger PB, Dunn AS, et al; American College of Chest Physicians. The perioperative management of antithrombotic therapy: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest 2008; 133(suppl 6):299S–339S.
- Douketis JD, Spyropoulos AC, Spencer FA, et al; American College of Chest Physicians. Perioperative management of antithrombotic therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e326S–e350S.
- Boehringer Ingelheim Pharmaceuticals, Inc. PRADAXA (dabigatran) package insert. http://bidocs.boehringer-ingelheim.com/BIWebAc-cess/ViewServlet.ser?docBase=renetnt&folderPath=/Prescribing%20Information/PIs/Pradaxa/Pradaxa.pdf. Accessed August 6, 2014.
- Levy JH, Faraoni D, Spring JL, Douketis JD, Samama CM. Managing new oral anticoagulants in the perioperative and intensive care unit setting. Anesthesiology 2013; 118:1466–1474.
- US Food and Drug Administration (FDA). FDA drug safety communication: update on the risk for serious bleeding events with the anticoagulant Pradaxa (dabigatran). www.fda.gov/drugs/drugsafety/ucm326580.htm. Accessed August 6, 2014.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- Schulman S, Kearon C, Kakkar AK, et al; RE-MEDY Trial Investigators. Extended use of dabigatran, warfarin, or placebo in venous thromboembolism. N Engl J Med 2013; 368:709–718.
- Eriksson BI, Dahl OE, Rosencher N, Büller HR, et al; RE-NOVATE Study Group. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement: a randomised, double-blind, non-inferiority trial. Lancet 2007; 370:949–956.
- Eriksson BI, Dahl OE, Rosencher N, et al; RE-MODEL Study Group. Oral dabigatran etexilate vs subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: the RE-MODEL randomized trial. J Thromb Haemost 2007; 5:2178–2185.
- Eikelboom JW, Connolly SJ, Brueckmann M, et al; RE-ALIGN Investigators. Dabigatran versus warfarin in patients with mechanical heart valves. N Engl J Med 2013; 369:1206–1214.
- Ageno W, Gallus AS, Wittkowsky A, Crowther M, Hylek EM, Palareti G; American College of Chest Physicians. Oral anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e44S–e88S.
- Blech S, Ebner T, Ludwig-Schwellinger E, Stangier J, Roth W. The metabolism and disposition of the oral direct thrombin inhibitor, dabigatran, in humans. Drug Metab Dispos 2008; 36:386–399.
- Janssen Pharmaceuticals, Inc. XARELTO (rivaroxaban) package insert. www.xareltohcp.com/about-xarelto/about-xarelto.html?utm_source=google&utm_medium=cpc&utm_campaign=Branded+-+Broad&utm_term=xarelto%20rivaroxaban&utm_content=Xarelto+Rivaroxaban|mkwid|sxSDxPb4m_dc|pcrid|34667840494. Accessed August 6, 2014.
- Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–391.
- EINSTEIN Investigators; Bauersachs R, Berkowitz SD, Brenner B, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med 2010; 363:2499–2510.
- EINSTEIN–PE Investigators; Büller HR, Prins MH, Lensin AW, et al. Oral rivaroxaban for the treatment of symptomatic pulmonary embolism. N Engl J Med 2012; 366:1287–1297.
- Eriksson BI, Borris LC, Friedman RJ, et al; RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med 2008; 358:2765–2775.
- Kakkar AK, Brenner B, Dahl OE, et al; RECORD2 Investigators. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet 2008; 372:31–39.
- Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med 2008; 358:2776–2786.
- Kubitza D, Becka M, Mueck W, et al. Effects of renal impairment on the pharmacokinetics, pharmacodynamics and safety of rivaroxaban, an oral, direct factor Xa inhibitor. Br J Clin Pharmacol 2010; 70:703–712.
- Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365:981–992.
- Bristol-Myers Squibb Company. ELIQUIS (apixaban) package insert. www.eliquis.com/index.aspx. Accessed August 6, 2014.
- Furie KL, Goldstein LB, Albers GW, et al; American Heart Association Stroke Council. Oral antithrombotic agents for the prevention of stroke in nonvalvular atrial fibrillation: a science advisory for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:3442–3453.
- ClinicalTrials.gov, US National Institutes of Health. PERIOP 2 - A Safety and Effectiveness Study of LMWH Bridging Therapy Versus Placebo Bridging Therapy for Patients on Long Term Warfarin and Require Temporary Interruption of Their Warfarin. http://clinicaltri-als.gov/show/NCT00432796. Accessed August 6, 2014.
- ClinicalTrials.gov, US National Institutes of Health. Effectiveness of Bridging Anticoagulation for Surgery (The BRIDGE Study). http://clinicaltrials.gov/ct2/show/NCT00786474. Accessed August 6, 2014.
- Birnie DH, Healey JS, Wells GA, et al; BRUISE CONTROL Investigators. Pacemaker or defibrillator surgery without interruption of anticoagulation. N Engl J Med 2013; 368:2084–2093.
- Oberweis BS, Nukala S, Rosenberg A, et al. Thrombotic and bleeding complications after orthopedic surgery. Am Heart J 2013; 165:427.e1–433.e1.
- Omran H, Bauersachs R, Rübenacker S, Goss F, Hammerstingl C. The HAS-BLED score predicts bleedings during bridging of chronic oral anticoagulation. Results from the national multicentre BNK Online bRiDging REgistRy (BORDER). Thromb Haemost 2012; 108:65–73.
- Pisters R, Lane DA, Nieuwlaaat R, de Vos CB, Crijns HJGM, Lip GYH. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation. The Euro Heart Survey. Chest 2010; 138:1093–1100.
- Kaatz S, Douketis JD, Zhou H, Gage BF, White RH. Risk of stroke after surgery in patients with and without chronic atrial fibrillation. J Thromb Haemost 2010; 8:884–890.
- van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost 2010; 103:1116–1127.
- Connolly G, Spyropoulos AC. Practical issues, limitations, and periprocedural management of the NOAC’s. J Thromb Thrombolysis 2013; 36:212–222.
- Spyropoulos AC, Douketis JD. How I treat anticoagulated patients undergoing an elective procedure or surgery. Blood 2012; 120:2954–2962.
- Kaatz S, Kouides PA, Garcia DA, et al. Guidance on the emergent reversal of oral thrombin and factor Xa inhibitors. Am J Hematol 2012; 87(suppl 1):S141–S145.
- Rowley CP, Bernard ML, Brabham WW, et al. Safety of continuous anticoagulation with dabigatran during implantation of cardiac rhythm devices. Am J Cardiol 2013; 111:1165–1168.
- Healey JS, Eikelboom J, Douketis J, et al; RE-LY Investigators. Periprocedural bleeding and thromboembolic events with dabigatran compared with warfarin: results from the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) randomized trial. Circulation 2012; 126:343–348.
- Sherwood MW, Douketis JD, Patel MR, et al; on behalf of the ROCKET AF Investigators. Outcomes of temporary interruption of rivaroxaban compared with warfarin in patients with nonvalvular atrial fibrillation: results from the Rivaroxaban Once Daily, Oral, Direct Factor Xa Inhibition Compared with Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation (ROCKET AF). Circulation 2014; 129:1850–1859.
- Patel MR, Hellkamp AS, Lokhnygina Y, et al. Outcomes of discontinuing rivaroxaban compared with warfarin in patients with nonvalvular atrial fibrillation: analysis from the ROCKET AF trial (Rivaroxaban Once-Daily, Oral, Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation). J Am Coll Cardiol 2013; 61:651–658.
- Reynolds MR. Discontinuation of rivaroxaban: filling in the gaps. J Am Coll Cardiol 2013; 61:659–660.
- Turpie AG, Kreutz R, Llau J, Norrving B, Haas S. Management consensus guidance for the use of rivaroxaban—an oral, direct factor Xa inhibitor. Thromb Haemost 2012; 108:876–886.
- Gallego P, Apostolakis S, Lip GY. Bridging evidence-based practice and practice-based evidence in periprocedural anticoagulation. Circulation 2012; 126:1573–1576.
- Sié P, Samama CM, Godier A, et al; Working Group on Perioperative Haemostasis. Surgery and invasive procedures in patients on long-term treatment with direct oral anticoagulants: thrombin or factor-Xa inhibitors. Recommendations of the Working Group on Perioperative Haemostasis and the French Study Group on Thrombosis and Haemostasis. Arch Cardiovasc Dis 2011; 104:669–676.
- King CS, Holley AB, Moores LK. Moving toward a more ideal anticoagulant: the oral direct thrombin and factor Xa inhibitors. Chest 2013; 143:1106–1116.
- Baglin T, Hillarp A, Tripodi A, Elalamy I, Buller H, Ageno W. Measuring oral direct inhibitors (ODIs) of thrombin and factor Xa: a recommendation from the Subcommittee on Control of Anticoagulation of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost 2013; 11:756–760.
- Baglin T, Keeling D, Kitchen S; British Committee for Standards in Haematology. Effects on routine coagulation screens and assessment of anticoagulant intensity in patients taking oral dabigatran or rivaroxaban: guidance from the British Committee for Standards in Haematology. Br J Haematol 2012; 159:427–429.
- Mani H, Kasper A, Lindhoff-Last E. Measuring the anticoagulant effects of target specific oral anticoagulants—reasons, methods and current limitations. J Thromb Thrombolysis 2013; 36:187–194.
- Hawes EM, Deal AM, Funk-Adcock D, et al. Performance of coagulation tests in patients on therapeutic doses of dabigatran: a cross-sectional pharmacodynamic study based on peak and trough plasma levels. J Thromb Haemost 2013; 11:1493–1502.
- Barrett YC, Wang Z, Frost C, Shenker A. Clinical laboratory measurement of direct factor Xa inhibitors: anti-Xa assay is preferable to prothrombin time assay. Thromb Haemost 2010; 104:1263–1271.
- Smythe MA, Fanikos J, Gulseth MP, et al. Rivaroxaban: practical considerations for ensuring safety and efficacy. Pharmacotherapy 2013; 33:1223–1245.
- Van Ryn J, Litzenburger T, Waterman A, et al. Dabigatran anticoagulant activity is neutralized by an antibody selective to dabigatran in in vitro and in vivo models. J Am Coll Cardiol 2011; 57:E1130.
- Sheffield W, Lambourne M, Bhakta V, Eltringham-Smith L, Arnold D, Crowther M. Active site-mutated thrombin S195A but not active site-blocked thrombin counteracts the anticoagulant activity of dabigatran in plasma. Abstract presented at the International Society of Thrombosis and Haemostasis 2013 Congress. http://onlinelibrary.wiley.com/doi/10.1111/jth.2013.11.issue-s2/issuetoc. Accessed August 6, 2014.
- Lu G, Luan P, Hollenbach SJ, et al. Reconstructed recombinant factor Xa as an antidote to reverse anticoagulation by factor Xa inhibitors (abstract). J Thromb Haemost 2009; 7(suppl 2):abstract OC-TH-107.
- Stangier J, Rathgen K, Stähle H, Mazur D. Influence of renal impairment on the pharmacokinetics and pharmacodynamics of oral dabigatran etexilate: an open-label, parallel-group, single-centre study. Clin Pharmacokinet 2010; 49:259–268.
- Singh T, Maw TT, Henry BL, et al. Extracorporeal therapy for dabigatran removal in the treatment of acute bleeding: a single center experience. Clin J Am Soc Nephrol 2013; 8:1533–1539.
- Kaatz S, Crowther M. Reversal of target-specific oral anticoagulants. J Thromb Thrombolysis 2013; 36:195–202.
- Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation 2011; 124:1573–1579.
- Sarode R, Milling TJ, Refaai MA, et al. Efficacy and safety of a 4-factor prothrombin complex concentrate in patients on vitamin K antagonists presenting with major bleeding: a randomized, plasma-controlled, phase IIIb study. Circulation 2013; 128:1234–1243.
KEY POINTS
- How long before surgery to stop a TSOAC depends on the bleeding risk of the procedure and the patient’s renal function.
- Perioperative bridging is generally unnecessary for patients on TSOACs.
- Routine coagulation assays such as the prothrombin time and activated partial thromboplastin time do not reliably reflect the degree of anticoagulation with TSOACs.
- There are no specific antidotes or standardized reversal strategies for TSOACs.
- TSOACs have a rapid onset of action and should only be restarted postoperatively once hemostasis has been confirmed.
Hospitalists and Sickle Cell Disease
Severe, disabling pain, often requiring opioids, is the most common medical presentation for children and adults with sickle cell disease (SCD), an autosomal recessive red blood cell disorder affecting those of African, Mediterranean, and Asian descent.1, 2 A genetically controlled hemoglobin alteration impairs oxygen binding, and enables polymerization of deoxy‐hemoglobin, resulting in, classically, sickle‐shaped erythrocytes3 and a complex cascade of ischemia and vaso‐occlusion in the microcirculation.4, 5
Dramatic gains in the treatment of SCD in childhood have resulted in markedly improved survival through adulthood.68 Thus, the need for adult SCD care is relatively new and rapidly growing. In 2005, approximately 70% of the nearly 80,000 US SCD hospitalizations occurred in adults versus children (Table 1). These hospitalizations occurred in the context of a poorly coordinated American health care system,9 despite the hopes raised by the Patient‐Centered Medical Home10 and the Chronic Care Model.11
| 2005 | 2008 | ||||||
|---|---|---|---|---|---|---|---|
| Total No. of Discharges | LOS | Total No. of Discharges | LOS | ||||
| |||||||
| All discharges | 79,187 | 100.00% | 5.3 | 70,121 | 100.00% | 5.4 | |
| Age group | <1 | 996 | 1.26% | 2.5 | 513 | 0.73% | 2.7 |
| 1‐17 | 23,134 | 29.21% | 3.9 | 13,754 | 19.62% | 3.8 | |
| 18‐44 | 48,168 | 60.83% | 6 | 48,021 | 68.48% | 5.8 | |
| 45‐64 | 6,527 | 8.24% | 6 | 7,543 | 10.76% | 5.6 | |
| 65‐84 | 281 | 0.35% | 6.4 | 221 | 0.32% | 5.6 | |
| Missing | 81 | 0.10% | 4 | 70 | 0.10% | 3.0 | |
Adults with SCD are vulnerable both because they are usually members of racial and ethnic minority groups, and because they have a Food and Drug Administration (FDA)‐defined orphan disease.12 They often do not receive the only FDA‐approved medication for SCD, life‐saving hydroxyurea,13 recommended for adults with homozygous sickle cell anemia (Hb SS) and sickle‐othalassemia (Hb SoThal).14 Young adults often fail to experience a smooth transition of care from children's hospitals, falling into a medical abyss.15
Therefore, increasingly, hospitalists are managing adults with SCD, rather than adult hematologyoncology, pain, or palliative care specialists. Adults with SCD experience negative opinions, bilateral lack of trust, and conflict in the doctorpatient relationship, frequently cited in studies of SCD adults and providers in the literature.16, 17
Evidence Base
General guidelines for SCD management have been published by the National Institutes of Health (NIH)18 and the Agency for Healthcare Policy and Research.19 But one of us (K.L.H.) found evidence lacking with regard to SCD pain management.20 Published guidelines on general pain management, such as the World Health Organization's Analgesic Ladder,21 do not address SCD. A Cochrane Review of pain management in SCD found only 9 randomized controlled trials, all with small numbers of patients, addressing acute SCD pain only.22 As well, American and British consensus SCD pain guidelines23, 24 admit, and subsequent publications emphasize,25, 26 the lack of evidence for what to do or not do for SCD pain management. At least 1 well‐done summary of the SCD evidence base intended for hospitalists has been published, but it focuses on management of issues other than pain.27
Motivations and Fears
It is not surprising then that hospitalists may bring great fear and apprehension with them into their care of SCD patients. One of us (W.R.S.), a general internist, has been called by his own and 3 other academic medical centers, 2 with active Federally‐funded SCD research programs, to address the problems of high‐utilizing adults with SCD, including counseling hospitalists frustrated with the management of pain in these patients.
Hospitalists may be motivated to provide efficient inpatient management (Table 2), and be aware of pain as the primary symptom of SCD inpatients. But they may carry knowledge gaps and biases into their relationships with SCD inpatients. They may fear opioid administration (opiophobia), loss of licensure or governmental reprisals because of high‐dose prescription of opioids, or may believe that SCD patients are more often addicted than most.17, 28 Consequently, more troublesome hospital stays may occur when patients are not rapidly and adequately titrated to appropriate analgesic doses, or when unnecessary deleterious side effects result from opioid and other analgesics. We therefore offer answers to frequently asked questions (FAQs) about pain management by hospitalists caring for adults with SCD. We address FAQs arising during the prototypical situationa patient with SCD admitted for a painful exacerbation, and little or no acute comorbidity. We refer the reader to the aforementioned articles and guidelines to address other treatment issues in adults with SCD.
| Principle | Obstacles |
|---|---|
| |
| Make appropriate management handoffs for patients coming from the ED to promote continuity of care and shorten hospitalization | Poor information systems and poor handoffs/continuity from ED management to hospital management |
| Get as much preexisting information about the patient as possible to inform acute care, avoid returns or further hospitalization | Patient may have no primary care physician or may underutilize primary care |
| Patient may misuse ED and hospital (as primary care source) | |
| Provide rapid and adequate analgesia | Ignorance of the differences between tolerance, physical dependence, addiction, and pseudoaddiction |
| No specific data on pharmacodynamics of opioid analgesics in sickle cell disease | |
| Don't lose licensure or arouse regulatory suspicion about prescribing patterns | Ignorance of DEA monitoring and laws governing appropriate vs inappropriate prescribing of opioids |
| Get the patient discharged as soon as medically appropriate | Difficulty assessing pain quality and intensity |
| Difficulty assessing/avoiding side effects of analgesics | |
| Difficulty determining appropriateness/timing of changes in analgesic dosing, discharge planning | |
| Make appropriate handoffs with the patient's usual source of continuity care (provide that source when necessary) to avoid returns or further hospitalization | Patient may have no primary care physician or may underutilize primary care physician |
| Patient may be mis/underprescribed analgesics by primary care physician | |
| Define and maintain appropriate roles for hospitalists vs physicians with sickle cell training, pain specialists, or other specialists | Inadequate adult system of care for sickle cell disease (no/paucity of specialty care) |
| Multiple prescribers of opioids | |
| Providers unwilling to care for or prescribe opioids for sickle cell patients | |
FAQS
-
Is there any objective way to tell when SCD patients really are in a crisis?
Although the term crisis is used as if it were an objectively definable biological entity, no one has proposed a standard definition of a crisis based on pain intensity level, clinical features, or biomarkers. Measures of vaso‐occlusion are correlated with ischemic pain, including pain that is often called a crisis.2932 However, neither ischemic pain from SCD, nor the underlying vaso‐occlusive cascade that is associated with this pain, is a sudden, present‐or‐absent phenomenon. Instead, these are continua that can be measured using pain scales or various biomarkers.
There is, however, correlative evidence of the intensity of SCD pain associated with various distinctive health states (admitted/not admitted, in crisis/not in crisis). The most visible measure of a crisis, health care utilization, was a strong predictor of mortality in the Cooperative Study of Sickle Cell Disease. Patients with 3 or more admissions per year had a lower 5‐year survival rate.33 In contrast, crisis in the landmark Pain in Sickle Cell Epidemiology Study (PiSCES) was self‐defined by patients.34 Despite being in pain on over half of their days on average, and despite a third of patients being in pain daily, most pain in PiSCES was not considered a crisis, and less than 5% of patients' days were spent in emergency departments (EDs) or hospitals. Ambulatory pain intensity reports were correlated with opioid use.35 A substantial minority (35%) of PiSCES patients made at least 3 ED visits per year. However, these high ED utilizers had worse laboratory values, more pain, more distress, and a lower quality of life.36
Importantly, sometimes adults with SCD may have severe comorbidities which may not be addressed or may be mistakenly managed as an acute vaso‐occlusive episode without further investigation or timely specialist consultation. Although pain is primarily the individual's chief complaint, any potential relationship between the presence of medical comorbidities and pain should be examined when patients are admitted.
-
How can one know when opioid dosages should be changed, or when SCD pain is appropriately controlled to allow discharge?
We recommend, as a standard of care, that SCD pain assessment and pain therapy be interwoven, despite a systematic review finding no evidence that directly linked the timing, frequency, or method of pain assessment with outcomes or safety in medical inpatients, and concluding that the safety and effectiveness of patient‐controlled analgesia (PCA) in medical patients had not been adequately studied.37 Hospitalists should focus the first 24 hours of inpatient SCD pain management on cycles of recurrent pain assessment and opioid dose titration as frequently as every 1 to 2 hours, to assure safe and rapidly efficacious analgesia. Pain intensity, duration, and character should be assessed directly. Intensity is often assessed using a visual analog scale (VAS) or numeric rating scale.38 Treating physicians should themselves directly assess pain during discussions of therapy with the patient, even though some assessment usually is done in hospitals during each nursing shift. Pain and pain relief can be assessed indirectly by monitoring opioid use.
We recommend PCA for inpatients with SCD, administered as an intermittent demand dose (patient must push a button) of opioid, with or without a background opioid constant infusion.39 We usually set the interval between doses, or lockout, to 6 to 10 minutes. Both the lockout and the sedation from delivered doses prevent patients from pushing the demand button repetitively to the point of overdose. Use of a low‐level constant infusion (basal) may sustain pain control during times when the patient is asleep, avoiding recrudescent pain and lost ground due to inadequate analgesia during rest. Alternatively, long‐acting oral opioids may be continued if already used at home, or newly introduced to provide adequate baseline pain control which is augmented by the demand dosing. Most PCA pumps monitor hourly opioid dose demand (number of pushes), as well as hourly doses delivered. Both hourly opioid dose demand and hourly dose‐per‐demand ratio are measures of PCA efficacy or futility. Pumps record this data, and can be interrogated at the patient's bedside for up to several days of prior use. Physicians should combine pump interrogation with direct pain assessment to guide demand‐dose titration. Demand doses should be increased to 1.5 to 2 times the previous demand dose after several hours of failed reduction of pain intensity and duration, and/or persistently futile dose‐per‐demand ratios.
PCA interrogation is also useful for conversion of parenteral opioids to oral opioids, as well as to guide the recommendation for discharge home. After the first 24‐48 hours of up‐titration, if opioid dose demand decreases concordantly with pain frequency and intensity, the demand dose may be safely decreased, and eventually daily PCA requirements may be summed and converted to oral medication using standard opioid dose conversion tables. At this point, physicians may use single measures or daily averages of directly assessed pain.
Routine PCA use in SCD is backed by some evidence.40, 41 But we find it important that patients be taught and encouraged to use the demand feature of PCA. Still, for various reasons, some patients do not use PCA pumps well. Discordant or unreliable assessments (eg, high pain intensity but low‐opioid demand doses during the same interval) may result, and PCA potentially may fail as a dosing strategy. Management is more difficult for these patients. One alternative dosing strategy is prescription of scheduled doses of a short‐acting opioid, attaching to each dose the order, patient may refuse. This is different than dosing as needed, and allows counts of dose refusals over an interval, analogous to PCA pump interrogation.
-
How much is too much opioid? Should one rely on side effects, or on requests for medicine, or is there a ceiling dose?
Addictionologists, pain specialists, oncologists, those involved in hospice care, and some hematologists caring for SCD patients agree that, in general, there should be no a priori dose limitations imposed on opioid prescribing for acute pain. Instead, titration of dose of opioid to pain relief is a central principle of acute pain management. Experts also agree that particular opioids carry particular side effects which warrant dose limitation, adjustments, or avoidance of that opioid altogether. A summary of opioids commonly used in SCD, along with warnings and implied dose limitations is found in Table 3.
For safety, it is important to assess the history of prior opioid use to recognize a patient who is not tolerant to opioids (see below, FAQ 4), to avoid mistakenly overdosing a patient using doses often required by tolerant patients. In lieu of a pre‐written, individualized opioid dosing plan in place for the patient, the patient may be the best source of information regarding preferred medication and tolerated doses.
The reader is referred to standard texts for a description of opioids, their pharmacokinetics and pharmacodynamics, and their addictive and abuse potential. The side‐effect profile of opioids is well‐known: nausea, vomiting, and itching frequently occur; hallucinations, respiratory suppression, and myoclonus occur infrequently.42 Meperidine may more readily cause central nervous system (CNS) dysfunction, including seizures, as compared to other commonly used opioids, because of its toxic metabolite nor‐meperidine. Use of meperidine is often avoided, especially use via PCA.43 Methadone may cause dysrhythmias, specificially corrected Q‐T interval (QTc) prolongation and torsades de pointes on an electrocardiogram, in doses above 200 mg per day.44 Some recommend baseline and yearly electrocardiogram monitoring when giving methadone chronically.
Recognizing the potential dangers of opioids, it is also reasonable to look for opioid‐sparing analgesic strategies. Non‐opioid analgesics such as ketorolac45 and adjuvants such as ketamine46 that are opioid‐sparing should be considered whenever feasible. Complementary and alternative therapies such as transcutaneous electrical nerve stimulation (TENS)47 have less evidence of effectiveness, but have limited risks and may be of use for some individuals.
-
What are the major signs of substance abuse (opioids, street drugs) in SCD patients already on opioids, and can a hospitalist judge those signs acutely and intervene appropriately?
Reports of underprescription of opioids in SCD have cited physician fear of abuse and addiction.48 A recent informal poll of adult sickle cell providers suggests policies vary on how potential abuse is monitored in ambulatory sickle cell patients. We note that physicians, especially upon meeting a patient for the first time, may be unable to reliably judge whether that patient is abusing opioids or street drugs. Both false‐positive and false‐negative diagnoses may be made.49 Repetitive reports of lost or stolen prescriptions or pill bottles, receipt of prescriptions from multiple providers, or repeated requests for early refills increase the suspicion of misuse or abuse, but are indirect evidence. Urine and serum monitoring may be useful, but may give incorrect information if misinterpreted or not conducted frequently enough to improve sensitivity.50
It is important to distinguish between tolerance, the decreased analgesic response over time to repeated doses of the same drug; physical dependence, the production of withdrawal upon abrupt discontinuation of an opioid agonist or administration of an antagonist; and addiction, the psychological dependence upon opioids. Tolerance may be misperceived as true addiction. Its earliest symptom is shortening of the duration of effective analgesia. In contrast, addiction may be manifested by dose escalation in the absence of an increased pain stimulus, or by use of opioids for purposes other than pain relief.51 These are not easily distinguished during a single patient encounter.
SCD patients' requests for specific opioid medications in specific doses, should not be taken as evidence of past or current abuse, but rather evidence of a well‐informed, self‐managing patient. Adults with SCD are clearly expected to be very knowledgeable about and tolerant to opioids if they have had a life of pain as a child, and will require higher doses of opioids than other patients treated by most hospitalists. The issue of medication abuse may be best handled in the ambulatory setting. Whenever possible, hospitalists should not rely only on data from the acute care setting to manage patients. Ambulatory providers may conduct random, unannounced urine and/or serum testing, as part of an opioid prescribing agreement that is written and filed in the patient's chart. Assays for prescribed opioids (especially long‐acting agents), as well as assays for common drugs of abuse, should be conducted. Comanagement with an addictionologist, psychiatrist, or psychologist should be considered in individuals suspected of opioid abuse.
We do not suggest routine urine drug test monitoring of all SCD patients unless routine monitoring is done as a policy for all patients on opioids. Though the prevalence of addiction may be higher in subpopulations of patients with pain,52 and though prescription of opioids, prescription drug abuse, and accidental deaths from prescribed opioids have risen exponentially in the last several years,53 in our experience and in the published literature, drug misuse/abuse among SCD patients is no worse than among patients with other illnesses.5456 However, pseudoaddiction, the appropriate seeking of needed opioids from multiple physicians because of uncontrolled pain and opioid underprescription, may well be prevalent in SCD,57 and may be mistaken for true addiction.
-
How can patients' readiness for discharge be assessed? What can be done for the patient who has lengthy and/or multiple hospitalizations or frequent ED visits?
The appropriate time for discharge in most patients is when they can manage their pain at home with oral opioids or less. Often, patients do not improve even after a few days of inpatient therapy.58 A typical pain episode may last much longer than the 6‐day average US hospital length of stay for a diagnosis of sickle cell crisis among 18‐44 year olds (Table 1).59 Patients may return and be readmitted.60, 61 But in the best cases, pain resolves or reverts to a usual chronic intensity level. As described in FAQ 2, daily or more frequent pain assessment is a bedrock for making discharge decisions. Patients well‐experienced in the use of pain intensity scales can report their usual pain intensity at home, and how close they are to their baseline pain intensity. Simply asking patients, Are you ready for discharge? is appropriate and may yield a surprising positive response. In a recent inpatient trial of PCA (manuscript in preparation), adult patients were admitted with a minimum pain intensity of 45 mm on a 100 mm horizontal VAS scale after treatment in the ED, and mean pain intensity of 76 mm 10 mm. All adults in this study were discharged with pain that was clinically significantly lower. Researchers have found a VAS change of 13.5 mm to be the minimum clinically significant change62 during treatment of vaso‐occlusive crisis.63
Unremitting pain despite appropriate titration of opioids and prolonged hospital stays suggests the need for comprehensive evaluation for medical and psychosocial comorbidities, as is done for other patients with chronic pain syndromes. If not already done, discussion with the patient's primary care provider may reveal factors impacting on persistent pain. Consultation with a hematologist, pain or palliative care specialist, or other provider familiar with SCD may prove helpful. Implementation of adjuvant therapies as discussed in FAQ 3 and adding long‐acting oral opioids to continue postdischarge may also help. Hyperalgesia, or heightened sensitivity to pain, is normal after acute tissue injury, but is now suspected in SCD as a long‐term neuropathic pain syndrome, as a consequence either of repeated painful crises or of chronic opioid therapy.2 Only some centers have specialists qualified to test for and diagnose neuropathic pain.64
Discharge planning should include identification of a source of outpatient follow‐up. Opioids prescribed at discharge should be sufficient to last at least until the first outpatient appointment, to avoid repeated ED or hospital visits. Communication with a primary care provider at discharge can enhance successful care transition. Otherwise, for patients without established providers, social workers and others may address barriers to follow‐up that frustrate both patient and provider.
| Opioid | Used Frequently (>20% of Patients) | How Used | Unique Side Effects and/or Dose Limitations |
|---|---|---|---|
| |||
| Short‐acting | |||
| Codeine | No | Inpatient, parenteral; Ambulatory, oral | |
| Oxycodone | Yes | Most commonly used ambulatory opioid | |
| Morphine | Yes | Most commonly used inpatient opioid | |
| Hydromorphone | Yes | Inpatient more than ambulatory | |
| Fentanyl | No | Inpatient, parenteral | Short‐acting |
| Hydrocodone | No | Ambulatory | |
| Meperedine | No | Avoided | Unpredictable seizure, coma, death |
| Propoxyphene | No | Ambulatory | |
| Tramadol | No | Ambulatory | |
| Long‐acting | |||
| Oxycodone | No | Ambulatory and as an oral basal in inpatients | Abuse potential from capsule manipulation |
| Morphine | Yes | Ambulatory and as an oral basal in inpatients; most commonly used long‐acting opioid | |
| Methadone | No | Ambulatory and as an oral basal in inpatients | Dose‐dependent prolongation of QTc, torsades de pointes |
| Fentanyl | No | Ambulatory and as a transdermal basal in inpatients | Abuse potential from transdermal patch manipulation |
Support for Hospitalists Managing Adults With Sickle Cell Disease
Beside the general advice on pain management in SCD mentioned above or found in the bibliography of this article, at long last, a group of adult practitioners skilled in the care of SCD has formed nationally. The Sickle Cell Adult Provider Network [
Ultimately, evidence and updated guidelines will be the best support for hospitalists and others managing pain in SCD. The hope is that SCD will receive the attention it deserves, so that practitioners and patients alike do not suffer continued pain from this disease or its management.
- .Sickle‐cell disease.Lancet.1997;350(9079):725–302.
- ,.Sickle‐cell pain: advances in epidemiology and etiology.Hematology Am Soc Hematol Educ Program.2010;409–415. PMID: 21239827.
- .Management of sickle cell disease.N Engl J Med.1999;340:1021–1030.
- ,,.A systems biology consideration of the vasculopathy of sickle cell anemia: the need for multi‐modality chemo‐prophylaxsis.Cardiovasc Hematol Disord Drug Targets.2009;9(4):271–292.
- ,,.Newer aspects of the pathophysiology of sickle cell disease vaso‐occlusion [review].Hemoglobin.2009;33(1):1–16.
- ,,,.National trends in the mortality of children with sickle cell disease, 1968 through 1992.Am J Public Health.1997;87(8):1317–1322.
- ,,, et al.Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography.N Engl J Med.1998;339:5–11.
- ,.Optimizing primary stroke prevention in sickle cell anemia (STOP 2) trial investigators. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease.N Engl J Med.2005;353(26):2769–2778.
- .Homeostasis without reserve—the risk of health system collapse.N Engl J Med.2002;347(24):1971–1975.
- The Advanced Medical Home: A Patient‐Centered, Physician‐Guided Model of Health Care [Policy Monograph].Philadelphia, PA:American College of Physicians;2006.
- ,,,,,.Quality improvement in chronic illness care: a collaborative approach.Jt Comm J Qual Improv.2001;27:63–80.
- Definition of Disease Prevalence for Therapies Qualifying Under the Orphan Drug Act. Subpart C, Designation of an Orphan Drug. Sec. 316.20. Content and format of a request for orphan‐drug designation. Available at: http://www.fda.gov/orphan/designat/prevalence.html. Accessed September 3,2008.
- ,,, et al.Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment.JAMA.2003;289:1645–1651.
- ,,,.Provider barriers to hydroxyurea use in adults with sickle cell disease: a survey of the Sickle Cell Disease Adult Provider Network.J Natl Med Assoc.2008;100(8):968–973.
- ,,,,.Transition from pediatric to adult care in sickle cell disease: establishing evidence‐based practice and directions for research.Am J Hematol.2011;86(1):116–120. PMID: 21061308.
- ,,,,,.Health perceptions and medical care opinions of inner‐city adults with sickle cell disease or asthma compared with those of their siblings.South Med J.1989;82(1):9–12.
- ,,,.Sickle cell‐related pain: perceptions of medical practitioners.J Pain Symptom Manage.1997;14(3):168–174.
- The Management of Sickle Cell Disease.4th ed. NIH Publication 2002–2117.Washington, DC:National Institutes of Health, National Heart, Lung, and Blood Institute, Division of Blood Diseases and Resources; June2002.
- Pain Management Guideline.Washington, DC:Agency for Health Care Policy and Research;1992.
- ,.An evidence‐based approach to the treatment of adults with sickle cell disease.Hematology Am Soc Hematol Educ Program.2005;58–65.
- World Health Organization: Cancer Pain Relief.Geneva, Switzerland:WHO,1986.
- ,.Pain management for sickle cell disease.Cochrane Database Syst Rev. April 19,2006;(2):CD003350.
- ,,;for the Guideline Committee.Guidelines for the Management of Acute and Chronic Pain in Sickle Cell Disease. APS Clinical Practice Guideline Series, No 1.Glenview, IL:American Pain Society;1999.
- .Guidelines for the management of the acute painful crisis of sickle cell disease.Br J Haematol.2003;120:744–752.
- ,,.Acute pain in children and adults with sickle cell disease: management in the absence of evidence‐based guidelines.Curr Opin Hematol.2009;16(3):173–178.
- ,,,.Opioids and the treatment of chronic pain: controversies, current status, and future directions.Exp Clin Psychopharmacol.2008;16(5):405–416.
- .A sickle cell primer.The Hospitalist.2006;10(10):39–41.
- .The barriers to adequate pain management with opioid analgesics.Semin Oncol.1993;20(2 suppl 1):1–5.
- ,,,,,.Vaso‐occlusion in children with sickle cell disease: clinical characteristics and biologic correlates.J Pediatr Hematol Oncol.2004;26:785–790. PMID: 15591896.
- ,,,,,.Plasma endothelin‐1, cytokine, and prostaglandin E2 levels in sickle cell disease and acute vaso‐occlusive sickle crisis.Blood.1998;92:2551–2555. PMID: 9746797.
- ,,,.Serum levels of substance P are elevated in patients with sickle cell disease and increase further during vaso‐occlusive crisis.Blood.1998;92:3148–3151. PMID: 9787150.
- ,,,,;for the CURAMA study group.Plasma concentrations of asymmetric dimethylarginine, an endogenous nitric oxide synthase inhibitor, are elevated in sickle cell patients but do not increase further during painful crisis.Am J Hematol. February 27,2008; PMID: 18383318
- ,,, et al.Pain in sickle cell disease: rates and risk factors.N Engl J Med.1991;325:11–16.
- ,,, et al.Understanding pain and improving management of sickle cell disease: the PiSCES Study.J Natl Med Assoc.2005;97(2):183–193.
- ,,, et al.Daily assessment of pain in adults with sickle cell disease.Ann Intern Med.2008;148(2):94–101.
- ,,, et al.Comparisons of high versus low emergency department utilizers in sickle cell disease.Ann Emerg Med.2009;53(5):587–593.
- ,.Assessment and management of acute pain in adult medical inpatients: a systematic review.Pain Med.2009;10(7):1183–1199. PMID: 19818030.
- .Scaling clinical pain and pain relief. In: Bromm B, ed.Pain Measurement in Man: Neorephysiological Correlates of Pain.New York:Elsevier Science Publishers,1984:389–396.
- .Patient‐controlled analgesia for acute pain.Clin J Pain.1989;5(suppl 1):S8–S15. PMID: 2520435.
- ,,,,.Patient‐controlled analgesia for sickle pain crisis in pediatric emergency department.Pediatr Emerg Care.2004;20:2–4.
- ,,.A comparison of two regimens of patient‐controlled analgesia for children with sickle cell disease.J Pediatr Nurs.1998;13:15–19.
- Narcotic analgesics, 2002 update.The DAWN Report;2004.
- ,.Meperidine is alive and well in the new millennium: evaluation of meperidine usage patterns and frequency of adverse drug reactions.Pharmacotherapy.2004;24:776–783.
- ,,.Methadone‐related torsades de pointes in a sickle cell patient treated for chronic pain.Am J Hematol.2005;78(4):316–317.
- ,,,,,.A pilot study on the efficacy of ketorolac plus tramadol infusion combined with erythrocytapheresis in the management of acute severe vaso‐occlusive crises and sickle cell pain.Haematologica.2004;89(11):1389–1391.
- ,,,.Use of low‐dose ketamine infusion for pediatric patients with sickle cell disease‐related pain: a case series.Clin J Pain.2010;26(2):163–167.
- ,,.Transcutaneous electrical nerve stimulation treatment of sickle cell pain crises.Acta Haematol.1988;80(2):99–102.
- ,,.Psychosocial aspects of sickle cell disease (SCD) in childhood and adolescence: a review.Br J Clin Psychol.1993;32 (pt 3):271–280.
- ,,,,.Aberrant drug‐taking behaviors and headache: patient versus physician report.Am J Health Behav.2006;30(5):475–482.
- .Current issues in sickle cell pain and its management [review].Hematology Am Soc Hematol Educ Program.2007;97–105.
- ,,,.Opioid Abuse. Available at: http://www.emedicine.com/med/topic1673.htm. Updated April 18, 2006. Accessed August 23,2006.
- .Assessment for addiction in pain‐treatment settings.Clin J Pain.2002;18(4 suppl):S28–S38.
- ,,,,,,.Association between opioid prescribing patterns and opioid overdose‐related deaths.JAMA.2011;305(13):1315–1321.
- .American Pain Society workshop on the management of sickle cell pain.Saint Louis, MO;1990.
- ,,.Multidisciplinary approach to pain management in sickle cell disease.Am J Pediatr Hematol Oncol.1982;4:328–333.
- ,,,,,.Pain relief in sickle cell crisis [letter].Lancet.1986;2:624–625.
- ,,,,.Understanding the causes of problematic pain management in sickle cell disease: evidence that pseudoaddiction plays a more important role than genuine analgesic dependence.J Pain Symptom Manage.2004;27(2):156–169.
- ,.Red blood cell changes during the evolution of the sickle cell painful crisis.Blood.1992;79:2154–2163.
- ,,.Postdischarge pain, functional limitations and impact on caregivers of children with sickle cell disease treated for painful events.Br J Haematol.2009;144(5):782–788.
- ,.Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance.Am J Hematol.2005;79:17–25.
- ,,,,.Acute care utilization and rehospitalizations for sickle cell disease.JAMA.2010;303(13):1288–1294.
- ,,, et al.Clinical significance of reported changes in pain severity.Ann Emerg Med.1996;27:485–489.
- ,,,,.Clinically significant differences in visual analogue scale in acute vasoocclusive sickle cell crisis.Hemoglobin.2007;31:427–432.
- ,,.Usefulness and limitations of quantitative sensory testing: clinical and research application in neuropathic pain states.Pain.2007;129:256–259.
Severe, disabling pain, often requiring opioids, is the most common medical presentation for children and adults with sickle cell disease (SCD), an autosomal recessive red blood cell disorder affecting those of African, Mediterranean, and Asian descent.1, 2 A genetically controlled hemoglobin alteration impairs oxygen binding, and enables polymerization of deoxy‐hemoglobin, resulting in, classically, sickle‐shaped erythrocytes3 and a complex cascade of ischemia and vaso‐occlusion in the microcirculation.4, 5
Dramatic gains in the treatment of SCD in childhood have resulted in markedly improved survival through adulthood.68 Thus, the need for adult SCD care is relatively new and rapidly growing. In 2005, approximately 70% of the nearly 80,000 US SCD hospitalizations occurred in adults versus children (Table 1). These hospitalizations occurred in the context of a poorly coordinated American health care system,9 despite the hopes raised by the Patient‐Centered Medical Home10 and the Chronic Care Model.11
| 2005 | 2008 | ||||||
|---|---|---|---|---|---|---|---|
| Total No. of Discharges | LOS | Total No. of Discharges | LOS | ||||
| |||||||
| All discharges | 79,187 | 100.00% | 5.3 | 70,121 | 100.00% | 5.4 | |
| Age group | <1 | 996 | 1.26% | 2.5 | 513 | 0.73% | 2.7 |
| 1‐17 | 23,134 | 29.21% | 3.9 | 13,754 | 19.62% | 3.8 | |
| 18‐44 | 48,168 | 60.83% | 6 | 48,021 | 68.48% | 5.8 | |
| 45‐64 | 6,527 | 8.24% | 6 | 7,543 | 10.76% | 5.6 | |
| 65‐84 | 281 | 0.35% | 6.4 | 221 | 0.32% | 5.6 | |
| Missing | 81 | 0.10% | 4 | 70 | 0.10% | 3.0 | |
Adults with SCD are vulnerable both because they are usually members of racial and ethnic minority groups, and because they have a Food and Drug Administration (FDA)‐defined orphan disease.12 They often do not receive the only FDA‐approved medication for SCD, life‐saving hydroxyurea,13 recommended for adults with homozygous sickle cell anemia (Hb SS) and sickle‐othalassemia (Hb SoThal).14 Young adults often fail to experience a smooth transition of care from children's hospitals, falling into a medical abyss.15
Therefore, increasingly, hospitalists are managing adults with SCD, rather than adult hematologyoncology, pain, or palliative care specialists. Adults with SCD experience negative opinions, bilateral lack of trust, and conflict in the doctorpatient relationship, frequently cited in studies of SCD adults and providers in the literature.16, 17
Evidence Base
General guidelines for SCD management have been published by the National Institutes of Health (NIH)18 and the Agency for Healthcare Policy and Research.19 But one of us (K.L.H.) found evidence lacking with regard to SCD pain management.20 Published guidelines on general pain management, such as the World Health Organization's Analgesic Ladder,21 do not address SCD. A Cochrane Review of pain management in SCD found only 9 randomized controlled trials, all with small numbers of patients, addressing acute SCD pain only.22 As well, American and British consensus SCD pain guidelines23, 24 admit, and subsequent publications emphasize,25, 26 the lack of evidence for what to do or not do for SCD pain management. At least 1 well‐done summary of the SCD evidence base intended for hospitalists has been published, but it focuses on management of issues other than pain.27
Motivations and Fears
It is not surprising then that hospitalists may bring great fear and apprehension with them into their care of SCD patients. One of us (W.R.S.), a general internist, has been called by his own and 3 other academic medical centers, 2 with active Federally‐funded SCD research programs, to address the problems of high‐utilizing adults with SCD, including counseling hospitalists frustrated with the management of pain in these patients.
Hospitalists may be motivated to provide efficient inpatient management (Table 2), and be aware of pain as the primary symptom of SCD inpatients. But they may carry knowledge gaps and biases into their relationships with SCD inpatients. They may fear opioid administration (opiophobia), loss of licensure or governmental reprisals because of high‐dose prescription of opioids, or may believe that SCD patients are more often addicted than most.17, 28 Consequently, more troublesome hospital stays may occur when patients are not rapidly and adequately titrated to appropriate analgesic doses, or when unnecessary deleterious side effects result from opioid and other analgesics. We therefore offer answers to frequently asked questions (FAQs) about pain management by hospitalists caring for adults with SCD. We address FAQs arising during the prototypical situationa patient with SCD admitted for a painful exacerbation, and little or no acute comorbidity. We refer the reader to the aforementioned articles and guidelines to address other treatment issues in adults with SCD.
| Principle | Obstacles |
|---|---|
| |
| Make appropriate management handoffs for patients coming from the ED to promote continuity of care and shorten hospitalization | Poor information systems and poor handoffs/continuity from ED management to hospital management |
| Get as much preexisting information about the patient as possible to inform acute care, avoid returns or further hospitalization | Patient may have no primary care physician or may underutilize primary care |
| Patient may misuse ED and hospital (as primary care source) | |
| Provide rapid and adequate analgesia | Ignorance of the differences between tolerance, physical dependence, addiction, and pseudoaddiction |
| No specific data on pharmacodynamics of opioid analgesics in sickle cell disease | |
| Don't lose licensure or arouse regulatory suspicion about prescribing patterns | Ignorance of DEA monitoring and laws governing appropriate vs inappropriate prescribing of opioids |
| Get the patient discharged as soon as medically appropriate | Difficulty assessing pain quality and intensity |
| Difficulty assessing/avoiding side effects of analgesics | |
| Difficulty determining appropriateness/timing of changes in analgesic dosing, discharge planning | |
| Make appropriate handoffs with the patient's usual source of continuity care (provide that source when necessary) to avoid returns or further hospitalization | Patient may have no primary care physician or may underutilize primary care physician |
| Patient may be mis/underprescribed analgesics by primary care physician | |
| Define and maintain appropriate roles for hospitalists vs physicians with sickle cell training, pain specialists, or other specialists | Inadequate adult system of care for sickle cell disease (no/paucity of specialty care) |
| Multiple prescribers of opioids | |
| Providers unwilling to care for or prescribe opioids for sickle cell patients | |
FAQS
-
Is there any objective way to tell when SCD patients really are in a crisis?
Although the term crisis is used as if it were an objectively definable biological entity, no one has proposed a standard definition of a crisis based on pain intensity level, clinical features, or biomarkers. Measures of vaso‐occlusion are correlated with ischemic pain, including pain that is often called a crisis.2932 However, neither ischemic pain from SCD, nor the underlying vaso‐occlusive cascade that is associated with this pain, is a sudden, present‐or‐absent phenomenon. Instead, these are continua that can be measured using pain scales or various biomarkers.
There is, however, correlative evidence of the intensity of SCD pain associated with various distinctive health states (admitted/not admitted, in crisis/not in crisis). The most visible measure of a crisis, health care utilization, was a strong predictor of mortality in the Cooperative Study of Sickle Cell Disease. Patients with 3 or more admissions per year had a lower 5‐year survival rate.33 In contrast, crisis in the landmark Pain in Sickle Cell Epidemiology Study (PiSCES) was self‐defined by patients.34 Despite being in pain on over half of their days on average, and despite a third of patients being in pain daily, most pain in PiSCES was not considered a crisis, and less than 5% of patients' days were spent in emergency departments (EDs) or hospitals. Ambulatory pain intensity reports were correlated with opioid use.35 A substantial minority (35%) of PiSCES patients made at least 3 ED visits per year. However, these high ED utilizers had worse laboratory values, more pain, more distress, and a lower quality of life.36
Importantly, sometimes adults with SCD may have severe comorbidities which may not be addressed or may be mistakenly managed as an acute vaso‐occlusive episode without further investigation or timely specialist consultation. Although pain is primarily the individual's chief complaint, any potential relationship between the presence of medical comorbidities and pain should be examined when patients are admitted.
-
How can one know when opioid dosages should be changed, or when SCD pain is appropriately controlled to allow discharge?
We recommend, as a standard of care, that SCD pain assessment and pain therapy be interwoven, despite a systematic review finding no evidence that directly linked the timing, frequency, or method of pain assessment with outcomes or safety in medical inpatients, and concluding that the safety and effectiveness of patient‐controlled analgesia (PCA) in medical patients had not been adequately studied.37 Hospitalists should focus the first 24 hours of inpatient SCD pain management on cycles of recurrent pain assessment and opioid dose titration as frequently as every 1 to 2 hours, to assure safe and rapidly efficacious analgesia. Pain intensity, duration, and character should be assessed directly. Intensity is often assessed using a visual analog scale (VAS) or numeric rating scale.38 Treating physicians should themselves directly assess pain during discussions of therapy with the patient, even though some assessment usually is done in hospitals during each nursing shift. Pain and pain relief can be assessed indirectly by monitoring opioid use.
We recommend PCA for inpatients with SCD, administered as an intermittent demand dose (patient must push a button) of opioid, with or without a background opioid constant infusion.39 We usually set the interval between doses, or lockout, to 6 to 10 minutes. Both the lockout and the sedation from delivered doses prevent patients from pushing the demand button repetitively to the point of overdose. Use of a low‐level constant infusion (basal) may sustain pain control during times when the patient is asleep, avoiding recrudescent pain and lost ground due to inadequate analgesia during rest. Alternatively, long‐acting oral opioids may be continued if already used at home, or newly introduced to provide adequate baseline pain control which is augmented by the demand dosing. Most PCA pumps monitor hourly opioid dose demand (number of pushes), as well as hourly doses delivered. Both hourly opioid dose demand and hourly dose‐per‐demand ratio are measures of PCA efficacy or futility. Pumps record this data, and can be interrogated at the patient's bedside for up to several days of prior use. Physicians should combine pump interrogation with direct pain assessment to guide demand‐dose titration. Demand doses should be increased to 1.5 to 2 times the previous demand dose after several hours of failed reduction of pain intensity and duration, and/or persistently futile dose‐per‐demand ratios.
PCA interrogation is also useful for conversion of parenteral opioids to oral opioids, as well as to guide the recommendation for discharge home. After the first 24‐48 hours of up‐titration, if opioid dose demand decreases concordantly with pain frequency and intensity, the demand dose may be safely decreased, and eventually daily PCA requirements may be summed and converted to oral medication using standard opioid dose conversion tables. At this point, physicians may use single measures or daily averages of directly assessed pain.
Routine PCA use in SCD is backed by some evidence.40, 41 But we find it important that patients be taught and encouraged to use the demand feature of PCA. Still, for various reasons, some patients do not use PCA pumps well. Discordant or unreliable assessments (eg, high pain intensity but low‐opioid demand doses during the same interval) may result, and PCA potentially may fail as a dosing strategy. Management is more difficult for these patients. One alternative dosing strategy is prescription of scheduled doses of a short‐acting opioid, attaching to each dose the order, patient may refuse. This is different than dosing as needed, and allows counts of dose refusals over an interval, analogous to PCA pump interrogation.
-
How much is too much opioid? Should one rely on side effects, or on requests for medicine, or is there a ceiling dose?
Addictionologists, pain specialists, oncologists, those involved in hospice care, and some hematologists caring for SCD patients agree that, in general, there should be no a priori dose limitations imposed on opioid prescribing for acute pain. Instead, titration of dose of opioid to pain relief is a central principle of acute pain management. Experts also agree that particular opioids carry particular side effects which warrant dose limitation, adjustments, or avoidance of that opioid altogether. A summary of opioids commonly used in SCD, along with warnings and implied dose limitations is found in Table 3.
For safety, it is important to assess the history of prior opioid use to recognize a patient who is not tolerant to opioids (see below, FAQ 4), to avoid mistakenly overdosing a patient using doses often required by tolerant patients. In lieu of a pre‐written, individualized opioid dosing plan in place for the patient, the patient may be the best source of information regarding preferred medication and tolerated doses.
The reader is referred to standard texts for a description of opioids, their pharmacokinetics and pharmacodynamics, and their addictive and abuse potential. The side‐effect profile of opioids is well‐known: nausea, vomiting, and itching frequently occur; hallucinations, respiratory suppression, and myoclonus occur infrequently.42 Meperidine may more readily cause central nervous system (CNS) dysfunction, including seizures, as compared to other commonly used opioids, because of its toxic metabolite nor‐meperidine. Use of meperidine is often avoided, especially use via PCA.43 Methadone may cause dysrhythmias, specificially corrected Q‐T interval (QTc) prolongation and torsades de pointes on an electrocardiogram, in doses above 200 mg per day.44 Some recommend baseline and yearly electrocardiogram monitoring when giving methadone chronically.
Recognizing the potential dangers of opioids, it is also reasonable to look for opioid‐sparing analgesic strategies. Non‐opioid analgesics such as ketorolac45 and adjuvants such as ketamine46 that are opioid‐sparing should be considered whenever feasible. Complementary and alternative therapies such as transcutaneous electrical nerve stimulation (TENS)47 have less evidence of effectiveness, but have limited risks and may be of use for some individuals.
-
What are the major signs of substance abuse (opioids, street drugs) in SCD patients already on opioids, and can a hospitalist judge those signs acutely and intervene appropriately?
Reports of underprescription of opioids in SCD have cited physician fear of abuse and addiction.48 A recent informal poll of adult sickle cell providers suggests policies vary on how potential abuse is monitored in ambulatory sickle cell patients. We note that physicians, especially upon meeting a patient for the first time, may be unable to reliably judge whether that patient is abusing opioids or street drugs. Both false‐positive and false‐negative diagnoses may be made.49 Repetitive reports of lost or stolen prescriptions or pill bottles, receipt of prescriptions from multiple providers, or repeated requests for early refills increase the suspicion of misuse or abuse, but are indirect evidence. Urine and serum monitoring may be useful, but may give incorrect information if misinterpreted or not conducted frequently enough to improve sensitivity.50
It is important to distinguish between tolerance, the decreased analgesic response over time to repeated doses of the same drug; physical dependence, the production of withdrawal upon abrupt discontinuation of an opioid agonist or administration of an antagonist; and addiction, the psychological dependence upon opioids. Tolerance may be misperceived as true addiction. Its earliest symptom is shortening of the duration of effective analgesia. In contrast, addiction may be manifested by dose escalation in the absence of an increased pain stimulus, or by use of opioids for purposes other than pain relief.51 These are not easily distinguished during a single patient encounter.
SCD patients' requests for specific opioid medications in specific doses, should not be taken as evidence of past or current abuse, but rather evidence of a well‐informed, self‐managing patient. Adults with SCD are clearly expected to be very knowledgeable about and tolerant to opioids if they have had a life of pain as a child, and will require higher doses of opioids than other patients treated by most hospitalists. The issue of medication abuse may be best handled in the ambulatory setting. Whenever possible, hospitalists should not rely only on data from the acute care setting to manage patients. Ambulatory providers may conduct random, unannounced urine and/or serum testing, as part of an opioid prescribing agreement that is written and filed in the patient's chart. Assays for prescribed opioids (especially long‐acting agents), as well as assays for common drugs of abuse, should be conducted. Comanagement with an addictionologist, psychiatrist, or psychologist should be considered in individuals suspected of opioid abuse.
We do not suggest routine urine drug test monitoring of all SCD patients unless routine monitoring is done as a policy for all patients on opioids. Though the prevalence of addiction may be higher in subpopulations of patients with pain,52 and though prescription of opioids, prescription drug abuse, and accidental deaths from prescribed opioids have risen exponentially in the last several years,53 in our experience and in the published literature, drug misuse/abuse among SCD patients is no worse than among patients with other illnesses.5456 However, pseudoaddiction, the appropriate seeking of needed opioids from multiple physicians because of uncontrolled pain and opioid underprescription, may well be prevalent in SCD,57 and may be mistaken for true addiction.
-
How can patients' readiness for discharge be assessed? What can be done for the patient who has lengthy and/or multiple hospitalizations or frequent ED visits?
The appropriate time for discharge in most patients is when they can manage their pain at home with oral opioids or less. Often, patients do not improve even after a few days of inpatient therapy.58 A typical pain episode may last much longer than the 6‐day average US hospital length of stay for a diagnosis of sickle cell crisis among 18‐44 year olds (Table 1).59 Patients may return and be readmitted.60, 61 But in the best cases, pain resolves or reverts to a usual chronic intensity level. As described in FAQ 2, daily or more frequent pain assessment is a bedrock for making discharge decisions. Patients well‐experienced in the use of pain intensity scales can report their usual pain intensity at home, and how close they are to their baseline pain intensity. Simply asking patients, Are you ready for discharge? is appropriate and may yield a surprising positive response. In a recent inpatient trial of PCA (manuscript in preparation), adult patients were admitted with a minimum pain intensity of 45 mm on a 100 mm horizontal VAS scale after treatment in the ED, and mean pain intensity of 76 mm 10 mm. All adults in this study were discharged with pain that was clinically significantly lower. Researchers have found a VAS change of 13.5 mm to be the minimum clinically significant change62 during treatment of vaso‐occlusive crisis.63
Unremitting pain despite appropriate titration of opioids and prolonged hospital stays suggests the need for comprehensive evaluation for medical and psychosocial comorbidities, as is done for other patients with chronic pain syndromes. If not already done, discussion with the patient's primary care provider may reveal factors impacting on persistent pain. Consultation with a hematologist, pain or palliative care specialist, or other provider familiar with SCD may prove helpful. Implementation of adjuvant therapies as discussed in FAQ 3 and adding long‐acting oral opioids to continue postdischarge may also help. Hyperalgesia, or heightened sensitivity to pain, is normal after acute tissue injury, but is now suspected in SCD as a long‐term neuropathic pain syndrome, as a consequence either of repeated painful crises or of chronic opioid therapy.2 Only some centers have specialists qualified to test for and diagnose neuropathic pain.64
Discharge planning should include identification of a source of outpatient follow‐up. Opioids prescribed at discharge should be sufficient to last at least until the first outpatient appointment, to avoid repeated ED or hospital visits. Communication with a primary care provider at discharge can enhance successful care transition. Otherwise, for patients without established providers, social workers and others may address barriers to follow‐up that frustrate both patient and provider.
| Opioid | Used Frequently (>20% of Patients) | How Used | Unique Side Effects and/or Dose Limitations |
|---|---|---|---|
| |||
| Short‐acting | |||
| Codeine | No | Inpatient, parenteral; Ambulatory, oral | |
| Oxycodone | Yes | Most commonly used ambulatory opioid | |
| Morphine | Yes | Most commonly used inpatient opioid | |
| Hydromorphone | Yes | Inpatient more than ambulatory | |
| Fentanyl | No | Inpatient, parenteral | Short‐acting |
| Hydrocodone | No | Ambulatory | |
| Meperedine | No | Avoided | Unpredictable seizure, coma, death |
| Propoxyphene | No | Ambulatory | |
| Tramadol | No | Ambulatory | |
| Long‐acting | |||
| Oxycodone | No | Ambulatory and as an oral basal in inpatients | Abuse potential from capsule manipulation |
| Morphine | Yes | Ambulatory and as an oral basal in inpatients; most commonly used long‐acting opioid | |
| Methadone | No | Ambulatory and as an oral basal in inpatients | Dose‐dependent prolongation of QTc, torsades de pointes |
| Fentanyl | No | Ambulatory and as a transdermal basal in inpatients | Abuse potential from transdermal patch manipulation |
Support for Hospitalists Managing Adults With Sickle Cell Disease
Beside the general advice on pain management in SCD mentioned above or found in the bibliography of this article, at long last, a group of adult practitioners skilled in the care of SCD has formed nationally. The Sickle Cell Adult Provider Network [
Ultimately, evidence and updated guidelines will be the best support for hospitalists and others managing pain in SCD. The hope is that SCD will receive the attention it deserves, so that practitioners and patients alike do not suffer continued pain from this disease or its management.
Severe, disabling pain, often requiring opioids, is the most common medical presentation for children and adults with sickle cell disease (SCD), an autosomal recessive red blood cell disorder affecting those of African, Mediterranean, and Asian descent.1, 2 A genetically controlled hemoglobin alteration impairs oxygen binding, and enables polymerization of deoxy‐hemoglobin, resulting in, classically, sickle‐shaped erythrocytes3 and a complex cascade of ischemia and vaso‐occlusion in the microcirculation.4, 5
Dramatic gains in the treatment of SCD in childhood have resulted in markedly improved survival through adulthood.68 Thus, the need for adult SCD care is relatively new and rapidly growing. In 2005, approximately 70% of the nearly 80,000 US SCD hospitalizations occurred in adults versus children (Table 1). These hospitalizations occurred in the context of a poorly coordinated American health care system,9 despite the hopes raised by the Patient‐Centered Medical Home10 and the Chronic Care Model.11
| 2005 | 2008 | ||||||
|---|---|---|---|---|---|---|---|
| Total No. of Discharges | LOS | Total No. of Discharges | LOS | ||||
| |||||||
| All discharges | 79,187 | 100.00% | 5.3 | 70,121 | 100.00% | 5.4 | |
| Age group | <1 | 996 | 1.26% | 2.5 | 513 | 0.73% | 2.7 |
| 1‐17 | 23,134 | 29.21% | 3.9 | 13,754 | 19.62% | 3.8 | |
| 18‐44 | 48,168 | 60.83% | 6 | 48,021 | 68.48% | 5.8 | |
| 45‐64 | 6,527 | 8.24% | 6 | 7,543 | 10.76% | 5.6 | |
| 65‐84 | 281 | 0.35% | 6.4 | 221 | 0.32% | 5.6 | |
| Missing | 81 | 0.10% | 4 | 70 | 0.10% | 3.0 | |
Adults with SCD are vulnerable both because they are usually members of racial and ethnic minority groups, and because they have a Food and Drug Administration (FDA)‐defined orphan disease.12 They often do not receive the only FDA‐approved medication for SCD, life‐saving hydroxyurea,13 recommended for adults with homozygous sickle cell anemia (Hb SS) and sickle‐othalassemia (Hb SoThal).14 Young adults often fail to experience a smooth transition of care from children's hospitals, falling into a medical abyss.15
Therefore, increasingly, hospitalists are managing adults with SCD, rather than adult hematologyoncology, pain, or palliative care specialists. Adults with SCD experience negative opinions, bilateral lack of trust, and conflict in the doctorpatient relationship, frequently cited in studies of SCD adults and providers in the literature.16, 17
Evidence Base
General guidelines for SCD management have been published by the National Institutes of Health (NIH)18 and the Agency for Healthcare Policy and Research.19 But one of us (K.L.H.) found evidence lacking with regard to SCD pain management.20 Published guidelines on general pain management, such as the World Health Organization's Analgesic Ladder,21 do not address SCD. A Cochrane Review of pain management in SCD found only 9 randomized controlled trials, all with small numbers of patients, addressing acute SCD pain only.22 As well, American and British consensus SCD pain guidelines23, 24 admit, and subsequent publications emphasize,25, 26 the lack of evidence for what to do or not do for SCD pain management. At least 1 well‐done summary of the SCD evidence base intended for hospitalists has been published, but it focuses on management of issues other than pain.27
Motivations and Fears
It is not surprising then that hospitalists may bring great fear and apprehension with them into their care of SCD patients. One of us (W.R.S.), a general internist, has been called by his own and 3 other academic medical centers, 2 with active Federally‐funded SCD research programs, to address the problems of high‐utilizing adults with SCD, including counseling hospitalists frustrated with the management of pain in these patients.
Hospitalists may be motivated to provide efficient inpatient management (Table 2), and be aware of pain as the primary symptom of SCD inpatients. But they may carry knowledge gaps and biases into their relationships with SCD inpatients. They may fear opioid administration (opiophobia), loss of licensure or governmental reprisals because of high‐dose prescription of opioids, or may believe that SCD patients are more often addicted than most.17, 28 Consequently, more troublesome hospital stays may occur when patients are not rapidly and adequately titrated to appropriate analgesic doses, or when unnecessary deleterious side effects result from opioid and other analgesics. We therefore offer answers to frequently asked questions (FAQs) about pain management by hospitalists caring for adults with SCD. We address FAQs arising during the prototypical situationa patient with SCD admitted for a painful exacerbation, and little or no acute comorbidity. We refer the reader to the aforementioned articles and guidelines to address other treatment issues in adults with SCD.
| Principle | Obstacles |
|---|---|
| |
| Make appropriate management handoffs for patients coming from the ED to promote continuity of care and shorten hospitalization | Poor information systems and poor handoffs/continuity from ED management to hospital management |
| Get as much preexisting information about the patient as possible to inform acute care, avoid returns or further hospitalization | Patient may have no primary care physician or may underutilize primary care |
| Patient may misuse ED and hospital (as primary care source) | |
| Provide rapid and adequate analgesia | Ignorance of the differences between tolerance, physical dependence, addiction, and pseudoaddiction |
| No specific data on pharmacodynamics of opioid analgesics in sickle cell disease | |
| Don't lose licensure or arouse regulatory suspicion about prescribing patterns | Ignorance of DEA monitoring and laws governing appropriate vs inappropriate prescribing of opioids |
| Get the patient discharged as soon as medically appropriate | Difficulty assessing pain quality and intensity |
| Difficulty assessing/avoiding side effects of analgesics | |
| Difficulty determining appropriateness/timing of changes in analgesic dosing, discharge planning | |
| Make appropriate handoffs with the patient's usual source of continuity care (provide that source when necessary) to avoid returns or further hospitalization | Patient may have no primary care physician or may underutilize primary care physician |
| Patient may be mis/underprescribed analgesics by primary care physician | |
| Define and maintain appropriate roles for hospitalists vs physicians with sickle cell training, pain specialists, or other specialists | Inadequate adult system of care for sickle cell disease (no/paucity of specialty care) |
| Multiple prescribers of opioids | |
| Providers unwilling to care for or prescribe opioids for sickle cell patients | |
FAQS
-
Is there any objective way to tell when SCD patients really are in a crisis?
Although the term crisis is used as if it were an objectively definable biological entity, no one has proposed a standard definition of a crisis based on pain intensity level, clinical features, or biomarkers. Measures of vaso‐occlusion are correlated with ischemic pain, including pain that is often called a crisis.2932 However, neither ischemic pain from SCD, nor the underlying vaso‐occlusive cascade that is associated with this pain, is a sudden, present‐or‐absent phenomenon. Instead, these are continua that can be measured using pain scales or various biomarkers.
There is, however, correlative evidence of the intensity of SCD pain associated with various distinctive health states (admitted/not admitted, in crisis/not in crisis). The most visible measure of a crisis, health care utilization, was a strong predictor of mortality in the Cooperative Study of Sickle Cell Disease. Patients with 3 or more admissions per year had a lower 5‐year survival rate.33 In contrast, crisis in the landmark Pain in Sickle Cell Epidemiology Study (PiSCES) was self‐defined by patients.34 Despite being in pain on over half of their days on average, and despite a third of patients being in pain daily, most pain in PiSCES was not considered a crisis, and less than 5% of patients' days were spent in emergency departments (EDs) or hospitals. Ambulatory pain intensity reports were correlated with opioid use.35 A substantial minority (35%) of PiSCES patients made at least 3 ED visits per year. However, these high ED utilizers had worse laboratory values, more pain, more distress, and a lower quality of life.36
Importantly, sometimes adults with SCD may have severe comorbidities which may not be addressed or may be mistakenly managed as an acute vaso‐occlusive episode without further investigation or timely specialist consultation. Although pain is primarily the individual's chief complaint, any potential relationship between the presence of medical comorbidities and pain should be examined when patients are admitted.
-
How can one know when opioid dosages should be changed, or when SCD pain is appropriately controlled to allow discharge?
We recommend, as a standard of care, that SCD pain assessment and pain therapy be interwoven, despite a systematic review finding no evidence that directly linked the timing, frequency, or method of pain assessment with outcomes or safety in medical inpatients, and concluding that the safety and effectiveness of patient‐controlled analgesia (PCA) in medical patients had not been adequately studied.37 Hospitalists should focus the first 24 hours of inpatient SCD pain management on cycles of recurrent pain assessment and opioid dose titration as frequently as every 1 to 2 hours, to assure safe and rapidly efficacious analgesia. Pain intensity, duration, and character should be assessed directly. Intensity is often assessed using a visual analog scale (VAS) or numeric rating scale.38 Treating physicians should themselves directly assess pain during discussions of therapy with the patient, even though some assessment usually is done in hospitals during each nursing shift. Pain and pain relief can be assessed indirectly by monitoring opioid use.
We recommend PCA for inpatients with SCD, administered as an intermittent demand dose (patient must push a button) of opioid, with or without a background opioid constant infusion.39 We usually set the interval between doses, or lockout, to 6 to 10 minutes. Both the lockout and the sedation from delivered doses prevent patients from pushing the demand button repetitively to the point of overdose. Use of a low‐level constant infusion (basal) may sustain pain control during times when the patient is asleep, avoiding recrudescent pain and lost ground due to inadequate analgesia during rest. Alternatively, long‐acting oral opioids may be continued if already used at home, or newly introduced to provide adequate baseline pain control which is augmented by the demand dosing. Most PCA pumps monitor hourly opioid dose demand (number of pushes), as well as hourly doses delivered. Both hourly opioid dose demand and hourly dose‐per‐demand ratio are measures of PCA efficacy or futility. Pumps record this data, and can be interrogated at the patient's bedside for up to several days of prior use. Physicians should combine pump interrogation with direct pain assessment to guide demand‐dose titration. Demand doses should be increased to 1.5 to 2 times the previous demand dose after several hours of failed reduction of pain intensity and duration, and/or persistently futile dose‐per‐demand ratios.
PCA interrogation is also useful for conversion of parenteral opioids to oral opioids, as well as to guide the recommendation for discharge home. After the first 24‐48 hours of up‐titration, if opioid dose demand decreases concordantly with pain frequency and intensity, the demand dose may be safely decreased, and eventually daily PCA requirements may be summed and converted to oral medication using standard opioid dose conversion tables. At this point, physicians may use single measures or daily averages of directly assessed pain.
Routine PCA use in SCD is backed by some evidence.40, 41 But we find it important that patients be taught and encouraged to use the demand feature of PCA. Still, for various reasons, some patients do not use PCA pumps well. Discordant or unreliable assessments (eg, high pain intensity but low‐opioid demand doses during the same interval) may result, and PCA potentially may fail as a dosing strategy. Management is more difficult for these patients. One alternative dosing strategy is prescription of scheduled doses of a short‐acting opioid, attaching to each dose the order, patient may refuse. This is different than dosing as needed, and allows counts of dose refusals over an interval, analogous to PCA pump interrogation.
-
How much is too much opioid? Should one rely on side effects, or on requests for medicine, or is there a ceiling dose?
Addictionologists, pain specialists, oncologists, those involved in hospice care, and some hematologists caring for SCD patients agree that, in general, there should be no a priori dose limitations imposed on opioid prescribing for acute pain. Instead, titration of dose of opioid to pain relief is a central principle of acute pain management. Experts also agree that particular opioids carry particular side effects which warrant dose limitation, adjustments, or avoidance of that opioid altogether. A summary of opioids commonly used in SCD, along with warnings and implied dose limitations is found in Table 3.
For safety, it is important to assess the history of prior opioid use to recognize a patient who is not tolerant to opioids (see below, FAQ 4), to avoid mistakenly overdosing a patient using doses often required by tolerant patients. In lieu of a pre‐written, individualized opioid dosing plan in place for the patient, the patient may be the best source of information regarding preferred medication and tolerated doses.
The reader is referred to standard texts for a description of opioids, their pharmacokinetics and pharmacodynamics, and their addictive and abuse potential. The side‐effect profile of opioids is well‐known: nausea, vomiting, and itching frequently occur; hallucinations, respiratory suppression, and myoclonus occur infrequently.42 Meperidine may more readily cause central nervous system (CNS) dysfunction, including seizures, as compared to other commonly used opioids, because of its toxic metabolite nor‐meperidine. Use of meperidine is often avoided, especially use via PCA.43 Methadone may cause dysrhythmias, specificially corrected Q‐T interval (QTc) prolongation and torsades de pointes on an electrocardiogram, in doses above 200 mg per day.44 Some recommend baseline and yearly electrocardiogram monitoring when giving methadone chronically.
Recognizing the potential dangers of opioids, it is also reasonable to look for opioid‐sparing analgesic strategies. Non‐opioid analgesics such as ketorolac45 and adjuvants such as ketamine46 that are opioid‐sparing should be considered whenever feasible. Complementary and alternative therapies such as transcutaneous electrical nerve stimulation (TENS)47 have less evidence of effectiveness, but have limited risks and may be of use for some individuals.
-
What are the major signs of substance abuse (opioids, street drugs) in SCD patients already on opioids, and can a hospitalist judge those signs acutely and intervene appropriately?
Reports of underprescription of opioids in SCD have cited physician fear of abuse and addiction.48 A recent informal poll of adult sickle cell providers suggests policies vary on how potential abuse is monitored in ambulatory sickle cell patients. We note that physicians, especially upon meeting a patient for the first time, may be unable to reliably judge whether that patient is abusing opioids or street drugs. Both false‐positive and false‐negative diagnoses may be made.49 Repetitive reports of lost or stolen prescriptions or pill bottles, receipt of prescriptions from multiple providers, or repeated requests for early refills increase the suspicion of misuse or abuse, but are indirect evidence. Urine and serum monitoring may be useful, but may give incorrect information if misinterpreted or not conducted frequently enough to improve sensitivity.50
It is important to distinguish between tolerance, the decreased analgesic response over time to repeated doses of the same drug; physical dependence, the production of withdrawal upon abrupt discontinuation of an opioid agonist or administration of an antagonist; and addiction, the psychological dependence upon opioids. Tolerance may be misperceived as true addiction. Its earliest symptom is shortening of the duration of effective analgesia. In contrast, addiction may be manifested by dose escalation in the absence of an increased pain stimulus, or by use of opioids for purposes other than pain relief.51 These are not easily distinguished during a single patient encounter.
SCD patients' requests for specific opioid medications in specific doses, should not be taken as evidence of past or current abuse, but rather evidence of a well‐informed, self‐managing patient. Adults with SCD are clearly expected to be very knowledgeable about and tolerant to opioids if they have had a life of pain as a child, and will require higher doses of opioids than other patients treated by most hospitalists. The issue of medication abuse may be best handled in the ambulatory setting. Whenever possible, hospitalists should not rely only on data from the acute care setting to manage patients. Ambulatory providers may conduct random, unannounced urine and/or serum testing, as part of an opioid prescribing agreement that is written and filed in the patient's chart. Assays for prescribed opioids (especially long‐acting agents), as well as assays for common drugs of abuse, should be conducted. Comanagement with an addictionologist, psychiatrist, or psychologist should be considered in individuals suspected of opioid abuse.
We do not suggest routine urine drug test monitoring of all SCD patients unless routine monitoring is done as a policy for all patients on opioids. Though the prevalence of addiction may be higher in subpopulations of patients with pain,52 and though prescription of opioids, prescription drug abuse, and accidental deaths from prescribed opioids have risen exponentially in the last several years,53 in our experience and in the published literature, drug misuse/abuse among SCD patients is no worse than among patients with other illnesses.5456 However, pseudoaddiction, the appropriate seeking of needed opioids from multiple physicians because of uncontrolled pain and opioid underprescription, may well be prevalent in SCD,57 and may be mistaken for true addiction.
-
How can patients' readiness for discharge be assessed? What can be done for the patient who has lengthy and/or multiple hospitalizations or frequent ED visits?
The appropriate time for discharge in most patients is when they can manage their pain at home with oral opioids or less. Often, patients do not improve even after a few days of inpatient therapy.58 A typical pain episode may last much longer than the 6‐day average US hospital length of stay for a diagnosis of sickle cell crisis among 18‐44 year olds (Table 1).59 Patients may return and be readmitted.60, 61 But in the best cases, pain resolves or reverts to a usual chronic intensity level. As described in FAQ 2, daily or more frequent pain assessment is a bedrock for making discharge decisions. Patients well‐experienced in the use of pain intensity scales can report their usual pain intensity at home, and how close they are to their baseline pain intensity. Simply asking patients, Are you ready for discharge? is appropriate and may yield a surprising positive response. In a recent inpatient trial of PCA (manuscript in preparation), adult patients were admitted with a minimum pain intensity of 45 mm on a 100 mm horizontal VAS scale after treatment in the ED, and mean pain intensity of 76 mm 10 mm. All adults in this study were discharged with pain that was clinically significantly lower. Researchers have found a VAS change of 13.5 mm to be the minimum clinically significant change62 during treatment of vaso‐occlusive crisis.63
Unremitting pain despite appropriate titration of opioids and prolonged hospital stays suggests the need for comprehensive evaluation for medical and psychosocial comorbidities, as is done for other patients with chronic pain syndromes. If not already done, discussion with the patient's primary care provider may reveal factors impacting on persistent pain. Consultation with a hematologist, pain or palliative care specialist, or other provider familiar with SCD may prove helpful. Implementation of adjuvant therapies as discussed in FAQ 3 and adding long‐acting oral opioids to continue postdischarge may also help. Hyperalgesia, or heightened sensitivity to pain, is normal after acute tissue injury, but is now suspected in SCD as a long‐term neuropathic pain syndrome, as a consequence either of repeated painful crises or of chronic opioid therapy.2 Only some centers have specialists qualified to test for and diagnose neuropathic pain.64
Discharge planning should include identification of a source of outpatient follow‐up. Opioids prescribed at discharge should be sufficient to last at least until the first outpatient appointment, to avoid repeated ED or hospital visits. Communication with a primary care provider at discharge can enhance successful care transition. Otherwise, for patients without established providers, social workers and others may address barriers to follow‐up that frustrate both patient and provider.
| Opioid | Used Frequently (>20% of Patients) | How Used | Unique Side Effects and/or Dose Limitations |
|---|---|---|---|
| |||
| Short‐acting | |||
| Codeine | No | Inpatient, parenteral; Ambulatory, oral | |
| Oxycodone | Yes | Most commonly used ambulatory opioid | |
| Morphine | Yes | Most commonly used inpatient opioid | |
| Hydromorphone | Yes | Inpatient more than ambulatory | |
| Fentanyl | No | Inpatient, parenteral | Short‐acting |
| Hydrocodone | No | Ambulatory | |
| Meperedine | No | Avoided | Unpredictable seizure, coma, death |
| Propoxyphene | No | Ambulatory | |
| Tramadol | No | Ambulatory | |
| Long‐acting | |||
| Oxycodone | No | Ambulatory and as an oral basal in inpatients | Abuse potential from capsule manipulation |
| Morphine | Yes | Ambulatory and as an oral basal in inpatients; most commonly used long‐acting opioid | |
| Methadone | No | Ambulatory and as an oral basal in inpatients | Dose‐dependent prolongation of QTc, torsades de pointes |
| Fentanyl | No | Ambulatory and as a transdermal basal in inpatients | Abuse potential from transdermal patch manipulation |
Support for Hospitalists Managing Adults With Sickle Cell Disease
Beside the general advice on pain management in SCD mentioned above or found in the bibliography of this article, at long last, a group of adult practitioners skilled in the care of SCD has formed nationally. The Sickle Cell Adult Provider Network [
Ultimately, evidence and updated guidelines will be the best support for hospitalists and others managing pain in SCD. The hope is that SCD will receive the attention it deserves, so that practitioners and patients alike do not suffer continued pain from this disease or its management.
- .Sickle‐cell disease.Lancet.1997;350(9079):725–302.
- ,.Sickle‐cell pain: advances in epidemiology and etiology.Hematology Am Soc Hematol Educ Program.2010;409–415. PMID: 21239827.
- .Management of sickle cell disease.N Engl J Med.1999;340:1021–1030.
- ,,.A systems biology consideration of the vasculopathy of sickle cell anemia: the need for multi‐modality chemo‐prophylaxsis.Cardiovasc Hematol Disord Drug Targets.2009;9(4):271–292.
- ,,.Newer aspects of the pathophysiology of sickle cell disease vaso‐occlusion [review].Hemoglobin.2009;33(1):1–16.
- ,,,.National trends in the mortality of children with sickle cell disease, 1968 through 1992.Am J Public Health.1997;87(8):1317–1322.
- ,,, et al.Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography.N Engl J Med.1998;339:5–11.
- ,.Optimizing primary stroke prevention in sickle cell anemia (STOP 2) trial investigators. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease.N Engl J Med.2005;353(26):2769–2778.
- .Homeostasis without reserve—the risk of health system collapse.N Engl J Med.2002;347(24):1971–1975.
- The Advanced Medical Home: A Patient‐Centered, Physician‐Guided Model of Health Care [Policy Monograph].Philadelphia, PA:American College of Physicians;2006.
- ,,,,,.Quality improvement in chronic illness care: a collaborative approach.Jt Comm J Qual Improv.2001;27:63–80.
- Definition of Disease Prevalence for Therapies Qualifying Under the Orphan Drug Act. Subpart C, Designation of an Orphan Drug. Sec. 316.20. Content and format of a request for orphan‐drug designation. Available at: http://www.fda.gov/orphan/designat/prevalence.html. Accessed September 3,2008.
- ,,, et al.Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment.JAMA.2003;289:1645–1651.
- ,,,.Provider barriers to hydroxyurea use in adults with sickle cell disease: a survey of the Sickle Cell Disease Adult Provider Network.J Natl Med Assoc.2008;100(8):968–973.
- ,,,,.Transition from pediatric to adult care in sickle cell disease: establishing evidence‐based practice and directions for research.Am J Hematol.2011;86(1):116–120. PMID: 21061308.
- ,,,,,.Health perceptions and medical care opinions of inner‐city adults with sickle cell disease or asthma compared with those of their siblings.South Med J.1989;82(1):9–12.
- ,,,.Sickle cell‐related pain: perceptions of medical practitioners.J Pain Symptom Manage.1997;14(3):168–174.
- The Management of Sickle Cell Disease.4th ed. NIH Publication 2002–2117.Washington, DC:National Institutes of Health, National Heart, Lung, and Blood Institute, Division of Blood Diseases and Resources; June2002.
- Pain Management Guideline.Washington, DC:Agency for Health Care Policy and Research;1992.
- ,.An evidence‐based approach to the treatment of adults with sickle cell disease.Hematology Am Soc Hematol Educ Program.2005;58–65.
- World Health Organization: Cancer Pain Relief.Geneva, Switzerland:WHO,1986.
- ,.Pain management for sickle cell disease.Cochrane Database Syst Rev. April 19,2006;(2):CD003350.
- ,,;for the Guideline Committee.Guidelines for the Management of Acute and Chronic Pain in Sickle Cell Disease. APS Clinical Practice Guideline Series, No 1.Glenview, IL:American Pain Society;1999.
- .Guidelines for the management of the acute painful crisis of sickle cell disease.Br J Haematol.2003;120:744–752.
- ,,.Acute pain in children and adults with sickle cell disease: management in the absence of evidence‐based guidelines.Curr Opin Hematol.2009;16(3):173–178.
- ,,,.Opioids and the treatment of chronic pain: controversies, current status, and future directions.Exp Clin Psychopharmacol.2008;16(5):405–416.
- .A sickle cell primer.The Hospitalist.2006;10(10):39–41.
- .The barriers to adequate pain management with opioid analgesics.Semin Oncol.1993;20(2 suppl 1):1–5.
- ,,,,,.Vaso‐occlusion in children with sickle cell disease: clinical characteristics and biologic correlates.J Pediatr Hematol Oncol.2004;26:785–790. PMID: 15591896.
- ,,,,,.Plasma endothelin‐1, cytokine, and prostaglandin E2 levels in sickle cell disease and acute vaso‐occlusive sickle crisis.Blood.1998;92:2551–2555. PMID: 9746797.
- ,,,.Serum levels of substance P are elevated in patients with sickle cell disease and increase further during vaso‐occlusive crisis.Blood.1998;92:3148–3151. PMID: 9787150.
- ,,,,;for the CURAMA study group.Plasma concentrations of asymmetric dimethylarginine, an endogenous nitric oxide synthase inhibitor, are elevated in sickle cell patients but do not increase further during painful crisis.Am J Hematol. February 27,2008; PMID: 18383318
- ,,, et al.Pain in sickle cell disease: rates and risk factors.N Engl J Med.1991;325:11–16.
- ,,, et al.Understanding pain and improving management of sickle cell disease: the PiSCES Study.J Natl Med Assoc.2005;97(2):183–193.
- ,,, et al.Daily assessment of pain in adults with sickle cell disease.Ann Intern Med.2008;148(2):94–101.
- ,,, et al.Comparisons of high versus low emergency department utilizers in sickle cell disease.Ann Emerg Med.2009;53(5):587–593.
- ,.Assessment and management of acute pain in adult medical inpatients: a systematic review.Pain Med.2009;10(7):1183–1199. PMID: 19818030.
- .Scaling clinical pain and pain relief. In: Bromm B, ed.Pain Measurement in Man: Neorephysiological Correlates of Pain.New York:Elsevier Science Publishers,1984:389–396.
- .Patient‐controlled analgesia for acute pain.Clin J Pain.1989;5(suppl 1):S8–S15. PMID: 2520435.
- ,,,,.Patient‐controlled analgesia for sickle pain crisis in pediatric emergency department.Pediatr Emerg Care.2004;20:2–4.
- ,,.A comparison of two regimens of patient‐controlled analgesia for children with sickle cell disease.J Pediatr Nurs.1998;13:15–19.
- Narcotic analgesics, 2002 update.The DAWN Report;2004.
- ,.Meperidine is alive and well in the new millennium: evaluation of meperidine usage patterns and frequency of adverse drug reactions.Pharmacotherapy.2004;24:776–783.
- ,,.Methadone‐related torsades de pointes in a sickle cell patient treated for chronic pain.Am J Hematol.2005;78(4):316–317.
- ,,,,,.A pilot study on the efficacy of ketorolac plus tramadol infusion combined with erythrocytapheresis in the management of acute severe vaso‐occlusive crises and sickle cell pain.Haematologica.2004;89(11):1389–1391.
- ,,,.Use of low‐dose ketamine infusion for pediatric patients with sickle cell disease‐related pain: a case series.Clin J Pain.2010;26(2):163–167.
- ,,.Transcutaneous electrical nerve stimulation treatment of sickle cell pain crises.Acta Haematol.1988;80(2):99–102.
- ,,.Psychosocial aspects of sickle cell disease (SCD) in childhood and adolescence: a review.Br J Clin Psychol.1993;32 (pt 3):271–280.
- ,,,,.Aberrant drug‐taking behaviors and headache: patient versus physician report.Am J Health Behav.2006;30(5):475–482.
- .Current issues in sickle cell pain and its management [review].Hematology Am Soc Hematol Educ Program.2007;97–105.
- ,,,.Opioid Abuse. Available at: http://www.emedicine.com/med/topic1673.htm. Updated April 18, 2006. Accessed August 23,2006.
- .Assessment for addiction in pain‐treatment settings.Clin J Pain.2002;18(4 suppl):S28–S38.
- ,,,,,,.Association between opioid prescribing patterns and opioid overdose‐related deaths.JAMA.2011;305(13):1315–1321.
- .American Pain Society workshop on the management of sickle cell pain.Saint Louis, MO;1990.
- ,,.Multidisciplinary approach to pain management in sickle cell disease.Am J Pediatr Hematol Oncol.1982;4:328–333.
- ,,,,,.Pain relief in sickle cell crisis [letter].Lancet.1986;2:624–625.
- ,,,,.Understanding the causes of problematic pain management in sickle cell disease: evidence that pseudoaddiction plays a more important role than genuine analgesic dependence.J Pain Symptom Manage.2004;27(2):156–169.
- ,.Red blood cell changes during the evolution of the sickle cell painful crisis.Blood.1992;79:2154–2163.
- ,,.Postdischarge pain, functional limitations and impact on caregivers of children with sickle cell disease treated for painful events.Br J Haematol.2009;144(5):782–788.
- ,.Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance.Am J Hematol.2005;79:17–25.
- ,,,,.Acute care utilization and rehospitalizations for sickle cell disease.JAMA.2010;303(13):1288–1294.
- ,,, et al.Clinical significance of reported changes in pain severity.Ann Emerg Med.1996;27:485–489.
- ,,,,.Clinically significant differences in visual analogue scale in acute vasoocclusive sickle cell crisis.Hemoglobin.2007;31:427–432.
- ,,.Usefulness and limitations of quantitative sensory testing: clinical and research application in neuropathic pain states.Pain.2007;129:256–259.
- .Sickle‐cell disease.Lancet.1997;350(9079):725–302.
- ,.Sickle‐cell pain: advances in epidemiology and etiology.Hematology Am Soc Hematol Educ Program.2010;409–415. PMID: 21239827.
- .Management of sickle cell disease.N Engl J Med.1999;340:1021–1030.
- ,,.A systems biology consideration of the vasculopathy of sickle cell anemia: the need for multi‐modality chemo‐prophylaxsis.Cardiovasc Hematol Disord Drug Targets.2009;9(4):271–292.
- ,,.Newer aspects of the pathophysiology of sickle cell disease vaso‐occlusion [review].Hemoglobin.2009;33(1):1–16.
- ,,,.National trends in the mortality of children with sickle cell disease, 1968 through 1992.Am J Public Health.1997;87(8):1317–1322.
- ,,, et al.Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography.N Engl J Med.1998;339:5–11.
- ,.Optimizing primary stroke prevention in sickle cell anemia (STOP 2) trial investigators. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease.N Engl J Med.2005;353(26):2769–2778.
- .Homeostasis without reserve—the risk of health system collapse.N Engl J Med.2002;347(24):1971–1975.
- The Advanced Medical Home: A Patient‐Centered, Physician‐Guided Model of Health Care [Policy Monograph].Philadelphia, PA:American College of Physicians;2006.
- ,,,,,.Quality improvement in chronic illness care: a collaborative approach.Jt Comm J Qual Improv.2001;27:63–80.
- Definition of Disease Prevalence for Therapies Qualifying Under the Orphan Drug Act. Subpart C, Designation of an Orphan Drug. Sec. 316.20. Content and format of a request for orphan‐drug designation. Available at: http://www.fda.gov/orphan/designat/prevalence.html. Accessed September 3,2008.
- ,,, et al.Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment.JAMA.2003;289:1645–1651.
- ,,,.Provider barriers to hydroxyurea use in adults with sickle cell disease: a survey of the Sickle Cell Disease Adult Provider Network.J Natl Med Assoc.2008;100(8):968–973.
- ,,,,.Transition from pediatric to adult care in sickle cell disease: establishing evidence‐based practice and directions for research.Am J Hematol.2011;86(1):116–120. PMID: 21061308.
- ,,,,,.Health perceptions and medical care opinions of inner‐city adults with sickle cell disease or asthma compared with those of their siblings.South Med J.1989;82(1):9–12.
- ,,,.Sickle cell‐related pain: perceptions of medical practitioners.J Pain Symptom Manage.1997;14(3):168–174.
- The Management of Sickle Cell Disease.4th ed. NIH Publication 2002–2117.Washington, DC:National Institutes of Health, National Heart, Lung, and Blood Institute, Division of Blood Diseases and Resources; June2002.
- Pain Management Guideline.Washington, DC:Agency for Health Care Policy and Research;1992.
- ,.An evidence‐based approach to the treatment of adults with sickle cell disease.Hematology Am Soc Hematol Educ Program.2005;58–65.
- World Health Organization: Cancer Pain Relief.Geneva, Switzerland:WHO,1986.
- ,.Pain management for sickle cell disease.Cochrane Database Syst Rev. April 19,2006;(2):CD003350.
- ,,;for the Guideline Committee.Guidelines for the Management of Acute and Chronic Pain in Sickle Cell Disease. APS Clinical Practice Guideline Series, No 1.Glenview, IL:American Pain Society;1999.
- .Guidelines for the management of the acute painful crisis of sickle cell disease.Br J Haematol.2003;120:744–752.
- ,,.Acute pain in children and adults with sickle cell disease: management in the absence of evidence‐based guidelines.Curr Opin Hematol.2009;16(3):173–178.
- ,,,.Opioids and the treatment of chronic pain: controversies, current status, and future directions.Exp Clin Psychopharmacol.2008;16(5):405–416.
- .A sickle cell primer.The Hospitalist.2006;10(10):39–41.
- .The barriers to adequate pain management with opioid analgesics.Semin Oncol.1993;20(2 suppl 1):1–5.
- ,,,,,.Vaso‐occlusion in children with sickle cell disease: clinical characteristics and biologic correlates.J Pediatr Hematol Oncol.2004;26:785–790. PMID: 15591896.
- ,,,,,.Plasma endothelin‐1, cytokine, and prostaglandin E2 levels in sickle cell disease and acute vaso‐occlusive sickle crisis.Blood.1998;92:2551–2555. PMID: 9746797.
- ,,,.Serum levels of substance P are elevated in patients with sickle cell disease and increase further during vaso‐occlusive crisis.Blood.1998;92:3148–3151. PMID: 9787150.
- ,,,,;for the CURAMA study group.Plasma concentrations of asymmetric dimethylarginine, an endogenous nitric oxide synthase inhibitor, are elevated in sickle cell patients but do not increase further during painful crisis.Am J Hematol. February 27,2008; PMID: 18383318
- ,,, et al.Pain in sickle cell disease: rates and risk factors.N Engl J Med.1991;325:11–16.
- ,,, et al.Understanding pain and improving management of sickle cell disease: the PiSCES Study.J Natl Med Assoc.2005;97(2):183–193.
- ,,, et al.Daily assessment of pain in adults with sickle cell disease.Ann Intern Med.2008;148(2):94–101.
- ,,, et al.Comparisons of high versus low emergency department utilizers in sickle cell disease.Ann Emerg Med.2009;53(5):587–593.
- ,.Assessment and management of acute pain in adult medical inpatients: a systematic review.Pain Med.2009;10(7):1183–1199. PMID: 19818030.
- .Scaling clinical pain and pain relief. In: Bromm B, ed.Pain Measurement in Man: Neorephysiological Correlates of Pain.New York:Elsevier Science Publishers,1984:389–396.
- .Patient‐controlled analgesia for acute pain.Clin J Pain.1989;5(suppl 1):S8–S15. PMID: 2520435.
- ,,,,.Patient‐controlled analgesia for sickle pain crisis in pediatric emergency department.Pediatr Emerg Care.2004;20:2–4.
- ,,.A comparison of two regimens of patient‐controlled analgesia for children with sickle cell disease.J Pediatr Nurs.1998;13:15–19.
- Narcotic analgesics, 2002 update.The DAWN Report;2004.
- ,.Meperidine is alive and well in the new millennium: evaluation of meperidine usage patterns and frequency of adverse drug reactions.Pharmacotherapy.2004;24:776–783.
- ,,.Methadone‐related torsades de pointes in a sickle cell patient treated for chronic pain.Am J Hematol.2005;78(4):316–317.
- ,,,,,.A pilot study on the efficacy of ketorolac plus tramadol infusion combined with erythrocytapheresis in the management of acute severe vaso‐occlusive crises and sickle cell pain.Haematologica.2004;89(11):1389–1391.
- ,,,.Use of low‐dose ketamine infusion for pediatric patients with sickle cell disease‐related pain: a case series.Clin J Pain.2010;26(2):163–167.
- ,,.Transcutaneous electrical nerve stimulation treatment of sickle cell pain crises.Acta Haematol.1988;80(2):99–102.
- ,,.Psychosocial aspects of sickle cell disease (SCD) in childhood and adolescence: a review.Br J Clin Psychol.1993;32 (pt 3):271–280.
- ,,,,.Aberrant drug‐taking behaviors and headache: patient versus physician report.Am J Health Behav.2006;30(5):475–482.
- .Current issues in sickle cell pain and its management [review].Hematology Am Soc Hematol Educ Program.2007;97–105.
- ,,,.Opioid Abuse. Available at: http://www.emedicine.com/med/topic1673.htm. Updated April 18, 2006. Accessed August 23,2006.
- .Assessment for addiction in pain‐treatment settings.Clin J Pain.2002;18(4 suppl):S28–S38.
- ,,,,,,.Association between opioid prescribing patterns and opioid overdose‐related deaths.JAMA.2011;305(13):1315–1321.
- .American Pain Society workshop on the management of sickle cell pain.Saint Louis, MO;1990.
- ,,.Multidisciplinary approach to pain management in sickle cell disease.Am J Pediatr Hematol Oncol.1982;4:328–333.
- ,,,,,.Pain relief in sickle cell crisis [letter].Lancet.1986;2:624–625.
- ,,,,.Understanding the causes of problematic pain management in sickle cell disease: evidence that pseudoaddiction plays a more important role than genuine analgesic dependence.J Pain Symptom Manage.2004;27(2):156–169.
- ,.Red blood cell changes during the evolution of the sickle cell painful crisis.Blood.1992;79:2154–2163.
- ,,.Postdischarge pain, functional limitations and impact on caregivers of children with sickle cell disease treated for painful events.Br J Haematol.2009;144(5):782–788.
- ,.Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance.Am J Hematol.2005;79:17–25.
- ,,,,.Acute care utilization and rehospitalizations for sickle cell disease.JAMA.2010;303(13):1288–1294.
- ,,, et al.Clinical significance of reported changes in pain severity.Ann Emerg Med.1996;27:485–489.
- ,,,,.Clinically significant differences in visual analogue scale in acute vasoocclusive sickle cell crisis.Hemoglobin.2007;31:427–432.
- ,,.Usefulness and limitations of quantitative sensory testing: clinical and research application in neuropathic pain states.Pain.2007;129:256–259.