User login
Quality in urine microscopy: The eyes of the beholder
The urine is the window to the kidney.This oft-repeated adage impresses upon medical students and residents the importance of urine microscopy in the evaluation of patients with renal disorders.
While this phrase is likely an adaptation of the idea in ancient times that the urine reflected on humors or the quality of the soul, the understanding of the relevance of urine findings to the state of the kidneys likely rests with the pioneers of urine microscopy. As reviewed by Fogazzi and Cameron,1,2 the origins of direct inspection of urine under a microscope lie in the 17th century, with industrious physicians who used rudimentary microscopes to identify basic structures in the urine and correlated them to clinical presentations.1 At first, only larger structures could be seen, mostly crystals in patients with nephrolithiasis. As microscopes advanced, smaller structures such as “corpuscles” and “cylinders” could be seen that described cells and casts.1
In correlating these findings to patient presentations, a rudimentary understanding of renal pathology evolved long before the advent of the kidney biopsy. Lipid droplets were seen1 in patients swollen from dropsy, and later known to have nephrotic syndromes. In 1872, Harley first described the altered morphology of dysmorphic red blood cells in patients with Bright disease or glomerulonephritis.1,3 In 1979, Birch and Fairley recognized that the presence of acanthocytes differentiated glomerular from nonglomerular hematuria.4
DYSMORPHIC RED BLOOD CELLS: TYPES AND SIGNIFICANCE


URINE MICROSCOPY: THE NEPHROLOGIST’S ROLE
The tools used in urine microscopy have advanced significantly since its advent. But not all advances have led to improved patient care. Laboratories have trained technicians to perform urine microscopy. Analyzers can identify basic urinary structures using algorithms to compare them against stored reference images. More important, urine microscopy has been categorized by accreditation and inspection bodies as a “test” rather than a physician-performed competency. As such, definitions of quality in urine microscopy have shifted from the application of urinary findings to the care of the patient to the reproducibility of identifying individual structures in ways that can be documented with quality checks performed by nonclinicians. And since the governing bodies require laboratories to adhere to burdensome procedures to maintain accreditation (eg, the US Food and Drug Administration’s Clinical Laboratory Improvement Amendments), many hospitals have closed nephrologist-based urine laboratories.
This would be acceptable if laboratory-generated reports provided information equivalent to that obtained by the nephrologist. But such reports rarely include anything beyond the most rudimentary findings. In these reports, the red blood cell is not differentiated as dysmorphic or monomorphic. All casts are granular. Crystals are often the highlight of the report, usually an incidental finding of little relevance. Phase contrast and polarization are never performed.
Despite the poor quality of data provided in these reports, because of increasing regulations and time restrictions, a dwindling number of nephrologists perform urine microscopy even at teaching institutions. In an informal 2009 survey of nephrology fellowship program directors, 79% of responding programs relied solely on lab-generated reports for microscopic findings (verbal communication, Perazella, 2017).
There is general concern among medical educators about the surrendering of the physical examination and other techniques to technology.7,8 However, in many cases, such changes may improve the ability to make a correct diagnosis. When performed properly, urine microscopy can help determine the need for kidney biopsy, differentiate causes of acute kidney injury, and help guide decisions about therapy. Perazella showed that urine microscopy could reliably differentiate acute tubular necrosis from prerenal azotemia.9 Further, the severity of findings on urine microscopy has been associated with worse renal outcomes.10 At our institution, nephrologist-performed urine microscopy resulted in a change in cause of acute kidney injury in 25% of cases and a concrete change in management in 12% of patients (unpublished data).
With this in mind, it is concerning that the only evidence in the literature on this topic demonstrated that laboratory-based urine microscopy is actually a hindrance to its underlying purpose in acute kidney injury, which is to help identify the cause of the injury. Tsai et al11 showed that nephrologists identified the cause of acute kidney injury correctly 90% of the time when they performed their own urine microscopy, but this dropped to 23% when they relied on a laboratory-generated report. Interestingly, knowing the patient’s clinical history when performing the microscopy was important, as the accuracy was 69% when a report of another nephrologist’s microscopy findings was used.11
APPLYING RESULTS TO THE PATIENT
The purpose of urine microscopy in clinical care is to identify and understand the findings as they apply to the patient. When viewed from this perspective, the renal patient is clearly best served when the nephrologist familiar with the case performs urine microscopy, rather than a technician or analyzer in remote parts of the hospital with no connection to the patient.
Advances in technology or streamlining of hospital services do not always produce improvements in patient care, and how we define quality is integral to identifying when this is the case. Quality checklists can serve as guides to safe patient care but should not replace clinical decision-making. Direct physician involvement with our patients has concrete benefits, whether taking a history, performing a physical examination, reviewing radiologic images, or looking at specimens such as urine. It allows us to experience the amazing pathophysiology of human illness and to understand the nuances unique to each of our patients.
But most important, it reinforces the need for the direct bond, both emotional and physical, between us as healers and our patients.
- Fogazzi GB, Cameron JS. Urinary microscopy from the seventeenth century to the present day. Kidney Int 1996; 50:1058–1068.
- Cameron JS. A history of urine microscopy. Clin Chem Lab Med 2015; 53(suppl 2):s1453–s1464.
- Harley G. The Urine and Its Derangements. London: J and A Churchill, 1872:178–179.
- Birch DF, Fairley K. Hematuria: glomerular or non-glomerular? Lancet 1979; 314:845–846.
- Köhler H, Wandel E, Brunck B. Acanthocyturia—a characteristic marker for glomerular bleeding. Kidney Int 1991; 40:115–120.
- Daza JL, De Rosa M, De Rosa G. Dysmorphic red blood cells. Cleve Clin J Med 2018; 85:12–13.
- Jauhar S. The demise of the physical exam. N Engl J Med 2006; 354:548–551.
- Mangione S. When the tail wags the dog: clinical skills in the age of technology. Cleve Clin J Med 2017; 84:278–280.
- Perazella MA, Coca SG, Kanbay M, Brewster UC, Parikh CR. Diagnostic value of urine microscopy for differential diagnosis of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol 2008; 3:1615–1619.
- Perazella MA, Coca SG, Hall IE, Iyanam U, Koraishy M, Parikh CR. Urine microscopy is associated with severity and worsening of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol 2010; 5:402–408.
- Tsai JJ, Yeun JY, Kumar VA, Don BR. Comparison and interpretation of urinalysis performed by a nephrologist versus a hospital-based clinical laboratory. Am J Kidney Dis 2005; 46:820–829.
Additional Reading
Fogazzi GB, Garigali G, Pirovano B, Muratore MT, Raimondi S, Berti S. How to improve the teaching of urine microscopy. Clin Chem Lab Med 2007; 45:407–412.
Fogazzi GB, Secchiero S. The role of nephrologists in teaching urinary sediment examination. Am J Kidney Dis 2006; 47:713.
Fogazzi GB, Verdesca S, Garigali G. Urinalysis: core curriculum 2008. Am J Kidney Dis 2008; 51:1052–1067.
The urine is the window to the kidney.This oft-repeated adage impresses upon medical students and residents the importance of urine microscopy in the evaluation of patients with renal disorders.
While this phrase is likely an adaptation of the idea in ancient times that the urine reflected on humors or the quality of the soul, the understanding of the relevance of urine findings to the state of the kidneys likely rests with the pioneers of urine microscopy. As reviewed by Fogazzi and Cameron,1,2 the origins of direct inspection of urine under a microscope lie in the 17th century, with industrious physicians who used rudimentary microscopes to identify basic structures in the urine and correlated them to clinical presentations.1 At first, only larger structures could be seen, mostly crystals in patients with nephrolithiasis. As microscopes advanced, smaller structures such as “corpuscles” and “cylinders” could be seen that described cells and casts.1
In correlating these findings to patient presentations, a rudimentary understanding of renal pathology evolved long before the advent of the kidney biopsy. Lipid droplets were seen1 in patients swollen from dropsy, and later known to have nephrotic syndromes. In 1872, Harley first described the altered morphology of dysmorphic red blood cells in patients with Bright disease or glomerulonephritis.1,3 In 1979, Birch and Fairley recognized that the presence of acanthocytes differentiated glomerular from nonglomerular hematuria.4
DYSMORPHIC RED BLOOD CELLS: TYPES AND SIGNIFICANCE
URINE MICROSCOPY: THE NEPHROLOGIST’S ROLE
The tools used in urine microscopy have advanced significantly since its advent. But not all advances have led to improved patient care. Laboratories have trained technicians to perform urine microscopy. Analyzers can identify basic urinary structures using algorithms to compare them against stored reference images. More important, urine microscopy has been categorized by accreditation and inspection bodies as a “test” rather than a physician-performed competency. As such, definitions of quality in urine microscopy have shifted from the application of urinary findings to the care of the patient to the reproducibility of identifying individual structures in ways that can be documented with quality checks performed by nonclinicians. And since the governing bodies require laboratories to adhere to burdensome procedures to maintain accreditation (eg, the US Food and Drug Administration’s Clinical Laboratory Improvement Amendments), many hospitals have closed nephrologist-based urine laboratories.
This would be acceptable if laboratory-generated reports provided information equivalent to that obtained by the nephrologist. But such reports rarely include anything beyond the most rudimentary findings. In these reports, the red blood cell is not differentiated as dysmorphic or monomorphic. All casts are granular. Crystals are often the highlight of the report, usually an incidental finding of little relevance. Phase contrast and polarization are never performed.
Despite the poor quality of data provided in these reports, because of increasing regulations and time restrictions, a dwindling number of nephrologists perform urine microscopy even at teaching institutions. In an informal 2009 survey of nephrology fellowship program directors, 79% of responding programs relied solely on lab-generated reports for microscopic findings (verbal communication, Perazella, 2017).
There is general concern among medical educators about the surrendering of the physical examination and other techniques to technology.7,8 However, in many cases, such changes may improve the ability to make a correct diagnosis. When performed properly, urine microscopy can help determine the need for kidney biopsy, differentiate causes of acute kidney injury, and help guide decisions about therapy. Perazella showed that urine microscopy could reliably differentiate acute tubular necrosis from prerenal azotemia.9 Further, the severity of findings on urine microscopy has been associated with worse renal outcomes.10 At our institution, nephrologist-performed urine microscopy resulted in a change in cause of acute kidney injury in 25% of cases and a concrete change in management in 12% of patients (unpublished data).
With this in mind, it is concerning that the only evidence in the literature on this topic demonstrated that laboratory-based urine microscopy is actually a hindrance to its underlying purpose in acute kidney injury, which is to help identify the cause of the injury. Tsai et al11 showed that nephrologists identified the cause of acute kidney injury correctly 90% of the time when they performed their own urine microscopy, but this dropped to 23% when they relied on a laboratory-generated report. Interestingly, knowing the patient’s clinical history when performing the microscopy was important, as the accuracy was 69% when a report of another nephrologist’s microscopy findings was used.11
APPLYING RESULTS TO THE PATIENT
The purpose of urine microscopy in clinical care is to identify and understand the findings as they apply to the patient. When viewed from this perspective, the renal patient is clearly best served when the nephrologist familiar with the case performs urine microscopy, rather than a technician or analyzer in remote parts of the hospital with no connection to the patient.
Advances in technology or streamlining of hospital services do not always produce improvements in patient care, and how we define quality is integral to identifying when this is the case. Quality checklists can serve as guides to safe patient care but should not replace clinical decision-making. Direct physician involvement with our patients has concrete benefits, whether taking a history, performing a physical examination, reviewing radiologic images, or looking at specimens such as urine. It allows us to experience the amazing pathophysiology of human illness and to understand the nuances unique to each of our patients.
But most important, it reinforces the need for the direct bond, both emotional and physical, between us as healers and our patients.
The urine is the window to the kidney.This oft-repeated adage impresses upon medical students and residents the importance of urine microscopy in the evaluation of patients with renal disorders.
While this phrase is likely an adaptation of the idea in ancient times that the urine reflected on humors or the quality of the soul, the understanding of the relevance of urine findings to the state of the kidneys likely rests with the pioneers of urine microscopy. As reviewed by Fogazzi and Cameron,1,2 the origins of direct inspection of urine under a microscope lie in the 17th century, with industrious physicians who used rudimentary microscopes to identify basic structures in the urine and correlated them to clinical presentations.1 At first, only larger structures could be seen, mostly crystals in patients with nephrolithiasis. As microscopes advanced, smaller structures such as “corpuscles” and “cylinders” could be seen that described cells and casts.1
In correlating these findings to patient presentations, a rudimentary understanding of renal pathology evolved long before the advent of the kidney biopsy. Lipid droplets were seen1 in patients swollen from dropsy, and later known to have nephrotic syndromes. In 1872, Harley first described the altered morphology of dysmorphic red blood cells in patients with Bright disease or glomerulonephritis.1,3 In 1979, Birch and Fairley recognized that the presence of acanthocytes differentiated glomerular from nonglomerular hematuria.4
DYSMORPHIC RED BLOOD CELLS: TYPES AND SIGNIFICANCE
URINE MICROSCOPY: THE NEPHROLOGIST’S ROLE
The tools used in urine microscopy have advanced significantly since its advent. But not all advances have led to improved patient care. Laboratories have trained technicians to perform urine microscopy. Analyzers can identify basic urinary structures using algorithms to compare them against stored reference images. More important, urine microscopy has been categorized by accreditation and inspection bodies as a “test” rather than a physician-performed competency. As such, definitions of quality in urine microscopy have shifted from the application of urinary findings to the care of the patient to the reproducibility of identifying individual structures in ways that can be documented with quality checks performed by nonclinicians. And since the governing bodies require laboratories to adhere to burdensome procedures to maintain accreditation (eg, the US Food and Drug Administration’s Clinical Laboratory Improvement Amendments), many hospitals have closed nephrologist-based urine laboratories.
This would be acceptable if laboratory-generated reports provided information equivalent to that obtained by the nephrologist. But such reports rarely include anything beyond the most rudimentary findings. In these reports, the red blood cell is not differentiated as dysmorphic or monomorphic. All casts are granular. Crystals are often the highlight of the report, usually an incidental finding of little relevance. Phase contrast and polarization are never performed.
Despite the poor quality of data provided in these reports, because of increasing regulations and time restrictions, a dwindling number of nephrologists perform urine microscopy even at teaching institutions. In an informal 2009 survey of nephrology fellowship program directors, 79% of responding programs relied solely on lab-generated reports for microscopic findings (verbal communication, Perazella, 2017).
There is general concern among medical educators about the surrendering of the physical examination and other techniques to technology.7,8 However, in many cases, such changes may improve the ability to make a correct diagnosis. When performed properly, urine microscopy can help determine the need for kidney biopsy, differentiate causes of acute kidney injury, and help guide decisions about therapy. Perazella showed that urine microscopy could reliably differentiate acute tubular necrosis from prerenal azotemia.9 Further, the severity of findings on urine microscopy has been associated with worse renal outcomes.10 At our institution, nephrologist-performed urine microscopy resulted in a change in cause of acute kidney injury in 25% of cases and a concrete change in management in 12% of patients (unpublished data).
With this in mind, it is concerning that the only evidence in the literature on this topic demonstrated that laboratory-based urine microscopy is actually a hindrance to its underlying purpose in acute kidney injury, which is to help identify the cause of the injury. Tsai et al11 showed that nephrologists identified the cause of acute kidney injury correctly 90% of the time when they performed their own urine microscopy, but this dropped to 23% when they relied on a laboratory-generated report. Interestingly, knowing the patient’s clinical history when performing the microscopy was important, as the accuracy was 69% when a report of another nephrologist’s microscopy findings was used.11
APPLYING RESULTS TO THE PATIENT
The purpose of urine microscopy in clinical care is to identify and understand the findings as they apply to the patient. When viewed from this perspective, the renal patient is clearly best served when the nephrologist familiar with the case performs urine microscopy, rather than a technician or analyzer in remote parts of the hospital with no connection to the patient.
Advances in technology or streamlining of hospital services do not always produce improvements in patient care, and how we define quality is integral to identifying when this is the case. Quality checklists can serve as guides to safe patient care but should not replace clinical decision-making. Direct physician involvement with our patients has concrete benefits, whether taking a history, performing a physical examination, reviewing radiologic images, or looking at specimens such as urine. It allows us to experience the amazing pathophysiology of human illness and to understand the nuances unique to each of our patients.
But most important, it reinforces the need for the direct bond, both emotional and physical, between us as healers and our patients.
- Fogazzi GB, Cameron JS. Urinary microscopy from the seventeenth century to the present day. Kidney Int 1996; 50:1058–1068.
- Cameron JS. A history of urine microscopy. Clin Chem Lab Med 2015; 53(suppl 2):s1453–s1464.
- Harley G. The Urine and Its Derangements. London: J and A Churchill, 1872:178–179.
- Birch DF, Fairley K. Hematuria: glomerular or non-glomerular? Lancet 1979; 314:845–846.
- Köhler H, Wandel E, Brunck B. Acanthocyturia—a characteristic marker for glomerular bleeding. Kidney Int 1991; 40:115–120.
- Daza JL, De Rosa M, De Rosa G. Dysmorphic red blood cells. Cleve Clin J Med 2018; 85:12–13.
- Jauhar S. The demise of the physical exam. N Engl J Med 2006; 354:548–551.
- Mangione S. When the tail wags the dog: clinical skills in the age of technology. Cleve Clin J Med 2017; 84:278–280.
- Perazella MA, Coca SG, Kanbay M, Brewster UC, Parikh CR. Diagnostic value of urine microscopy for differential diagnosis of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol 2008; 3:1615–1619.
- Perazella MA, Coca SG, Hall IE, Iyanam U, Koraishy M, Parikh CR. Urine microscopy is associated with severity and worsening of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol 2010; 5:402–408.
- Tsai JJ, Yeun JY, Kumar VA, Don BR. Comparison and interpretation of urinalysis performed by a nephrologist versus a hospital-based clinical laboratory. Am J Kidney Dis 2005; 46:820–829.
Additional Reading
Fogazzi GB, Garigali G, Pirovano B, Muratore MT, Raimondi S, Berti S. How to improve the teaching of urine microscopy. Clin Chem Lab Med 2007; 45:407–412.
Fogazzi GB, Secchiero S. The role of nephrologists in teaching urinary sediment examination. Am J Kidney Dis 2006; 47:713.
Fogazzi GB, Verdesca S, Garigali G. Urinalysis: core curriculum 2008. Am J Kidney Dis 2008; 51:1052–1067.
- Fogazzi GB, Cameron JS. Urinary microscopy from the seventeenth century to the present day. Kidney Int 1996; 50:1058–1068.
- Cameron JS. A history of urine microscopy. Clin Chem Lab Med 2015; 53(suppl 2):s1453–s1464.
- Harley G. The Urine and Its Derangements. London: J and A Churchill, 1872:178–179.
- Birch DF, Fairley K. Hematuria: glomerular or non-glomerular? Lancet 1979; 314:845–846.
- Köhler H, Wandel E, Brunck B. Acanthocyturia—a characteristic marker for glomerular bleeding. Kidney Int 1991; 40:115–120.
- Daza JL, De Rosa M, De Rosa G. Dysmorphic red blood cells. Cleve Clin J Med 2018; 85:12–13.
- Jauhar S. The demise of the physical exam. N Engl J Med 2006; 354:548–551.
- Mangione S. When the tail wags the dog: clinical skills in the age of technology. Cleve Clin J Med 2017; 84:278–280.
- Perazella MA, Coca SG, Kanbay M, Brewster UC, Parikh CR. Diagnostic value of urine microscopy for differential diagnosis of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol 2008; 3:1615–1619.
- Perazella MA, Coca SG, Hall IE, Iyanam U, Koraishy M, Parikh CR. Urine microscopy is associated with severity and worsening of acute kidney injury in hospitalized patients. Clin J Am Soc Nephrol 2010; 5:402–408.
- Tsai JJ, Yeun JY, Kumar VA, Don BR. Comparison and interpretation of urinalysis performed by a nephrologist versus a hospital-based clinical laboratory. Am J Kidney Dis 2005; 46:820–829.
Additional Reading
Fogazzi GB, Garigali G, Pirovano B, Muratore MT, Raimondi S, Berti S. How to improve the teaching of urine microscopy. Clin Chem Lab Med 2007; 45:407–412.
Fogazzi GB, Secchiero S. The role of nephrologists in teaching urinary sediment examination. Am J Kidney Dis 2006; 47:713.
Fogazzi GB, Verdesca S, Garigali G. Urinalysis: core curriculum 2008. Am J Kidney Dis 2008; 51:1052–1067.
Anemia of chronic kidney disease: Treat it, but not too aggressively
Anemia is a frequent complication of chronic kidney disease, occurring in over 90% of patients receiving renal replacement therapy. It is associated with significant morbidity and mortality. While its pathogenesis is typically multifactorial, the predominant cause is failure of the kidneys to produce enough endogenous erythropoietin. The clinical approval of recombinant human erythropoietin in 1989 dramatically changed the treatment of anemia of chronic kidney disease, but randomized controlled trials yielded disappointing results when erythropoiesis-stimulating agents (ESAs) were used to raise hemoglobin to normal levels.
This article reviews the epidemiology and pathophysiology of anemia of chronic kidney disease and discusses the complicated and conflicting evidence regarding its treatment.
DEFINITION AND PREVALENCE
Anemia is defined as a hemoglobin concentration less than 13.0 g/dL for men and less than 12.0 g/dL for premenopausal women.1 It is more common in patients with impaired kidney function, especially when the glomerular filtration rate (GFR) falls below 60 mL/min. It is rare at GFRs higher than 80 mL/min,2 but as the GFR falls, the severity of the anemia worsens3 and its prevalence increases: almost 90% of patients with a GFR less than 30 mL/min are anemic.4
RENAL ANEMIA IS ASSOCIATED WITH BAD OUTCOMES
Anemia in chronic kidney disease is independently associated with risk of death. It is also an all-cause mortality multiplier, ie, it magnifies the risk of death from other disease states.5
In observational studies, anemia was associated with faster progression of left ventricular hypertrophy, inflammation, and increased myocardial and peripheral oxygen demand, thereby leading to worse cardiac outcomes with increased risk of myocardial infarction, coronary revascularization, and readmission for heart failure.6–8 Anemia is also associated with fatigue, depression, reduced exercise tolerance, stroke, and increased risk of rehospitalization.9–13
RENAL ANEMIA IS MULTIFACTORIAL
Anemia of chronic kidney disease is typically attributed to the decrease of erythropoietin production that accompanies the fall in GFR. However, the process is multifactorial, with several other contributing factors: absolute and functional iron deficiency, folate and vitamin B12 deficiencies, reduced red blood cell life span, and suppression of erythropoiesis by the uremic milieu.14
While it was once thought that chronic kidney disease leads to loss of erythropoietin-producing cells, it is now known that downregulation of hypoxia-inducible factor (HIF; a transcription factor) is at least partially responsible for the decrease in erythropoietin production15,16 and that this downregulation is reversible (see below).
ERYTHROPOIETIN, IRON, AND RED BLOOD CELLS
Erythropoietin production is triggered by hypoxia, mediated by HIF
Erythropoietin is produced primarily in the deep cortex and outer medulla of the kidneys by a special population of peritubular interstitial cells.17 The parenchymal cells of the liver also produce erythropoietin, but much less.18
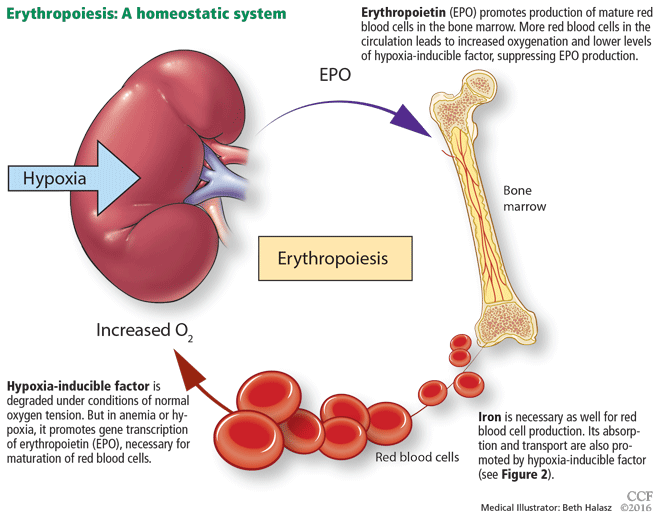
The rate of renal erythropoietin synthesis is determined by tissue oxygenation rather than by renal blood flow; production increases as the hemoglobin concentration drops and the arterial oxygen tension decreases (Figure 1).19
The gene for erythropoietin is located on chromosome 7 and is regulated by HIF. HIF molecules are composed of an alpha subunit, which is unstable at high Po2, and a beta subunit, constitutively present in the nucleus.20
In hypoxic conditions, the HIF dimer is transcriptionally active and binds to specific DNA recognition sequences called hypoxia-response elements. Gene transcription is upregulated, leading to increased production of erythropoietin.21
Under normal oxygen tension, on the other hand, the proline residue of the HIF alpha subunit is hydroxylated. The hydroxylated HIF alpha subunit is then degraded by proteasomal ubiquitylation, which is mediated by the von Hippel-Lindau tumor-suppressor gene pVHL.22 Degradation of HIF alpha prevents formation of the HIF heterodimers. HIF therefore cannot bind to the hypoxia-response elements, and erythropoietin gene transcription does not occur.23
Thus, in states of hypoxia, erythropoietin production is upregulated, whereas with normal oxygen tension, production is downregulated.
Erythropoietin is essential for terminal maturation of erythrocytes
Erythropoietin is essential for terminal maturation of erythrocytes.24 It is thought to stimulate the growth of erythrogenic progenitors: burst-forming units-erythroid (BFU-E) and colony-forming units-erythroid (CFU-E). In the absence of erythropoietin, BFU-E and CFU-E fail to differentiate into mature erythrocytes.25
Binding of erythropoietin to its receptor sets off a series of downstream signals, the most important being the signal transducer and activator of transcription 5 (STAT5). In animal studies, STAT5 was found to inhibit apoptosis through the early induction of an antiapoptotic gene, Bcl-xL.26
Iron metabolism is controlled by several proteins
Iron is characterized by its capacity to accept or donate electrons. This unique property makes it a crucial element in many biochemical reactions such as enzymatic activity, DNA synthesis, oxygen transport, and cell respiration.
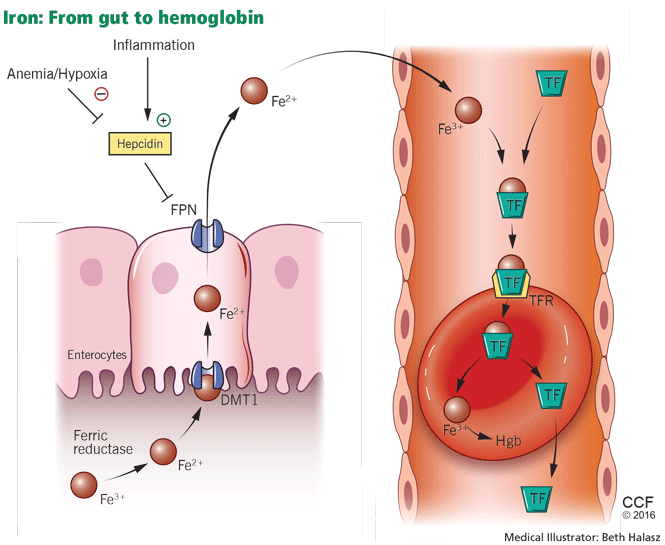
Iron metabolism is under the control of several proteins that play different roles in its absorption, recycling, and loss (Figure 2).27
Dietary iron exists primarily in its poorly soluble trivalent ferric form (Fe3+), and it needs to be reduced to its soluble divalent ferrous form (Fe2+) by ferric reductase to be absorbed. Ferrous iron is taken up at the apical side of enterocytes by a divalent metal transporter (DMT1) and is transported across the brush border.28
To enter the circulation, iron has to be transported across the basolateral membrane by a transporter called ferroportin.29 Ferroportin is also found in placental syncitiotrophoblasts, where it transfers iron from mother to fetus, and in macrophages, where it allows recycling of iron scavenged from damaged cells back into the circulation.30 Upon its release, the ferrous iron is oxidized to the ferric form and loaded onto transferrin. This oxidation process involves hephaestin, a homologue of the ferroxidase ceruloplasmin.31
In the plasma, iron is bound to transferrin, and under normal circumstances one-third of transferrin is saturated with iron.32 Transferrin receptors are present on most cells but are most dense on erythroid precursors. Each transferrin receptor can bind two transferrin molecules. After binding to transferrin, the transferrin receptor is endocytosed, and the iron is released into acidified vacuoles. The transferrin-receptor complex is then recycled to the surface.33
Ferritin is the cellular storage protein for iron, and it can store up to 4,500 atoms of iron within its spherical cavity.34 The serum level of ferritin reflects overall storage, with 1 ng/mL of ferritin indicating 10 mg of total iron stores.35 Ferritin is also an acute-phase reactant, and plasma levels can increase in inflammatory states such as infection or malignancy. As such, elevated ferritin does not necessarily indicate elevated iron stores.
Iron is lost in sweat, shed skin cells, and sloughed intestinal mucosal cells. However, there is no specific mechanism of iron excretion from the human body. Thus, iron is mainly regulated at the level of intestinal absorption. The iron exporter ferroportin is upregulated by the amount of available iron and is degraded by hepcidin.36
Hepcidin is a small cysteine-rich cationic peptide that is primarily produced in the liver, with some minor production also occurring in the kidneys.37 Transcription of the gene encoding hepcidin is downregulated by anemia and hypoxia and upregulated by inflammation and elevated iron levels.38 Transcription of hepcidin leads to degradation of ferroportin and a decrease in intestinal iron absorption. On the other hand, anemia and hypoxia inhibit hepcidin transcription, which allows ferroportin to facilitate intestinal iron absorption.
TREATMENT OF RENAL ANEMIA
Early enthusiasm for erythropoietin agents
Androgens started to be used to treat anemia of end-stage renal disease in 1970,39,40 and before the advent of recombinant human erythropoietin, they were a mainstay of nontransfusional therapy for anemic patients on dialysis.
The approval of recombinant human erythropoietin in 1989 drastically shifted the treatment of renal anemia. While the initial goal of treating anemia of chronic kidney disease with erythropoietin was to prevent blood transfusions,41 subsequent studies showed that the benefits might be far greater. Indeed, an initial observational trial showed that erythropoiesis-stimulating agents (ESAs) were associated with improved quality of life,42 improved neurocognitive function,43,44 and even cost savings.45 The benefits also extended to major outcomes such as regression of left ventricular hypertrophy,46 improvement in New York Heart Association class and cardiac function,47 fewer hospitalizations,48 and even reduction of cardiovascular mortality rates.49
As a result, ESA use gained popularity, and by 2006 an estimated 90% of dialysis patients were receiving these agents.50 The target and achieved hemoglobin levels also increased, with mean hemoglobin levels in hemodialysis patients being raised from 9.7 to 12 g/dL.51
Disappointing results in clinical trials of ESAs to normalize hemoglobin
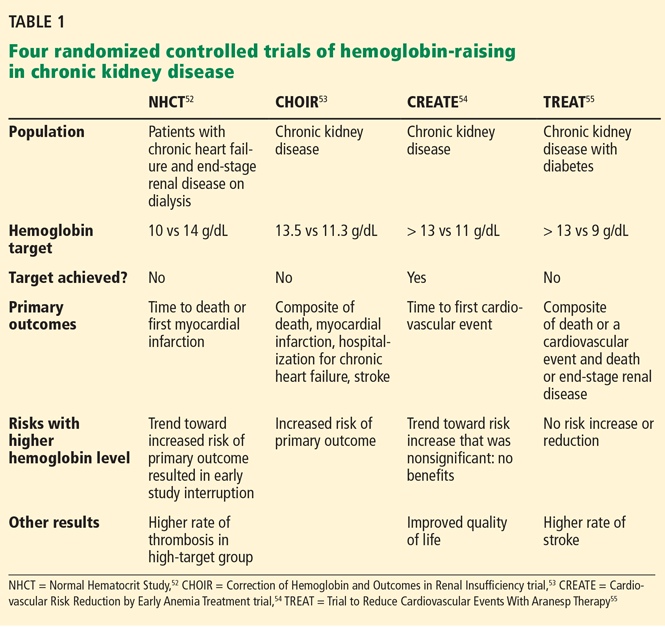
To prospectively study the effects of normalized hemoglobin targets, four randomized controlled trials were conducted (Table 1):
- The Normal Hematocrit Study (NHCT)52
- The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial53
- The Cardiovascular Risk Reduction by Early Anemia Treatment (CREATE) trial54
- The Trial to Reduce Cardiovascular Events With Aranesp Therapy (TREAT).55
These trials randomized patients to either higher “normal-range” hemoglobin targets or to lower target hemoglobin levels.
Their findings were disappointing and raised several red flags about excessive use of ESAs. The trials found no benefit in higher hemoglobin targets, and in fact, some of them demonstrated harm in patients randomized to higher targets. Notably, higher hemoglobin targets were associated with significant side effects such as access-site thrombosis,52 strokes,55 and possibly cardiovascular events.54,55 Only the CREATE trial was able to demonstrate a quality-of-life benefit for the high-target group.54
It remains unclear whether these adverse events were from the therapy itself or from an increased morbidity burden in the treated patients. Erythropoietin use is associated with hypertension,56 thought to be related to endothelin-mediated vasoconstriction.57 In our experience, this is most evident when hemoglobin levels are normalized with ESA therapy. Cycling of erythropoietin levels between extreme levels can lead to vascular remodeling, which may also be related to its cardiovascular effects.57
A noticeable finding in several of these trials was that patients failed to achieve the higher hemoglobin target despite the use of very high doses of ESA. Reanalysis of data from the CHOIR and CREATE trials showed that the patients who had worse outcomes were more likely to have required very high doses without achieving their target hemoglobin.58,59 Indeed, patients who achieved the higher target hemoglobin levels, usually at lower ESA doses, had better outcomes. This suggested that the need for a higher dose was associated with poorer outcomes, either as a marker of comorbidity or due to yet undocumented side effects of such high doses.
General approach to therapy
Before attributing anemia to chronic kidney disease, a thorough evaluation should be conducted to look for any reversible process that could be contributing to the anemia.
The causes of anemia are numerous and beyond the scope of this review. However, among the common causes of anemia in chronic kidney disease are deficiencies of iron, vitamin B12, and folate. Therefore, guidelines recommend checking iron, vitamin B12, and folate levels in the initial evaluation of anemia.60
Iron deficiency in particular is very common in chronic kidney disease patients and is present in nearly all dialysis patients.61 Hemodialysis patients are estimated to lose 1 to 3 g of iron per year as a result of blood loss in the dialysis circuit and increased iron utilization secondary to ESA therapy.62
However, in contrast to the general population, in which the upper limits of normal for iron indices are well defined, high serum ferritin levels appear to be poorly predictive of hemoglobin responsiveness in dialysis patients.63,64 Thus, the cutoffs that define iron responsiveness are much higher than standard definitions for iron deficiency.65,66 The Dialysis Patients’ Response to IV Iron With Elevated Ferritin (DRIVE) study showed that dialysis patients benefit from intravenous iron therapy even if their ferritin is as high as 1,200 ng/mL, provided their transferrin saturation is below 30%.67
Of note, erythropoietin levels cannot be used to distinguish renal anemia from other causes of anemia. Indeed, patients with renal failure may have “relative erythropoietin deficiency,” ie, “normal” erythropoietin levels that are actually too low in view of the degree of anemia.68,69 In addition to the decreased production capacity by the kidney, there appears to be a component of resistance to the action of erythropoietin in the bone marrow.
For these reasons, there is no erythropoietin level that can be considered “inadequate” or defining of renal anemia. Thus, measuring erythropoietin levels is not routinely recommended in the evaluation of renal anemia.
Two ESA preparations
The two ESAs that have traditionally been used in the treatment of renal anemia are recombinant human erythropoietin and darbepoietin alfa. They appear to be equivalent in terms of safety and efficacy.70 However, darbepoietin alfa has more sialic acid molecules, giving it a higher potency and longer half-life and allowing for less-frequent injections.71,72
In nondialysis patients, recombinant human erythropoietin is typically given every 1 to 2 weeks, whereas darbepoietin alfa can be given every 2 to 4 weeks. In dialysis patients, recombinant human erythropoietin is typically given 3 times per week with every dialysis treatment, while darbepoietin alfa is given once a week.
Target hemoglobin levels: ≤ 11.5 g/dL
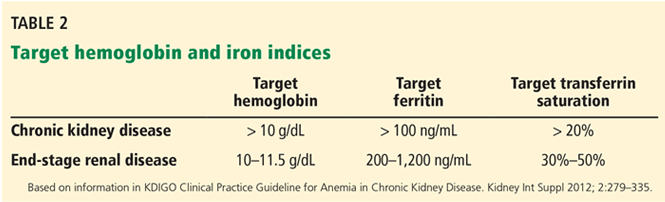
In light of the four trials described in Table 1, the international Kidney Disease: Improving Global Outcomes (KDIGO) guidelines60 recommend the following (Table 2):
For patients with chronic kidney disease who are not on dialysis, ESA therapy should not be initiated if the hemoglobin level is higher than 10 g/dL. If the hemoglobin level is lower than 10 g/dL, ESA therapy can be initiated, but the decision needs to be individualized based on the rate of fall of hemoglobin concentration, prior response to iron therapy, the risk of needing a transfusion, the risks related to ESA therapy, and the presence of symptoms attributable to anemia.
For patients on dialysis, ESA therapy should be used when the hemoglobin level is between 9 and 10 g/dL to avoid having the hemoglobin fall below 9 g/dL.
In all adult patients, ESAs should not be used to intentionally increase the hemoglobin level above 13 g/dL but rather to maintain levels no higher than 11.5 g/dL. This target is based on the observation that adverse outcomes were associated with ESA use with hemoglobin targets higher than 13 g/dL (Table 1).
Target iron levels
Regarding iron stores, the guidelines recommend the following:
For adult patients with chronic kidney disease who are not on dialysis, iron should be given to keep transferrin saturation above 20% and ferritin above 100 ng/mL. Transferrin saturation should not exceed 30%, and ferritin levels should not exceed 500 ng/mL.
For adult patients on dialysis, iron should be given to maintain transferrin saturation above 30% and ferritin above 200 ng/mL.
The upper limits of ferritin and transferrin saturation are somewhat controversial, as the safety of intentionally maintaining respective levels greater than 30% and 500 ng/mL has been studied in very few patients. Transferrin saturation should in general not exceed 50%.
High ferritin levels are associated with higher death rates, but whether elevation of ferritin levels is a marker of excessive iron administration rather than a nonspecific acute-phase reactant is not clear. The 2006 guidelines60 cited upper ferritin limits of 500 to 800 ng/mL. However, the more recent DRIVE trial67 showed that patients with ferritin levels of 500 to 1,200 ng/mL will respond to intravenous administration of iron with an increase in their hemoglobin levels. This has led many clinicians to adopt a higher ferritin limit of 1,200 ng/mL.
Hemosiderosis, or excess iron deposition, was a known consequence of frequent transfusions in patients with end-stage renal disease before ESA therapy was available. However, there have been no documented cases of clinical iron overload from iron therapy using current guidelines.73
These algorithms are nuanced, and the benefit of giving intravenous iron should always be weighed against the risks of short-term acute toxicity and infection. Treatment of renal anemia not only requires in-depth knowledge of the topic, but also familiarity with the patient’s specific situation. As such, it is not recommended that clinicians unfamiliar with the treatment of renal anemia manage its treatment.
PARTICULAR CIRCUMSTANCES
Inflammation and ESA resistance
While ESAs are effective in treating anemia in many cases, in many patients the anemia fails to respond. This is of particular importance, since ESA hyporesponsiveness has been found to be a powerful predictor of cardiovascular events and death.74 It is unclear, however, whether high doses of ESA are inherently toxic or whether hyporesponsiveness is a marker of adverse outcomes related to comorbidities.
KDIGO defines initial hyporesponsiveness as having no increase in hemoglobin concentration after the first month of appropriate weight-based dosing, and acquired hyporesponsiveness as requiring two increases in ESA doses up to 50% beyond the dose at which the patient had originally been stable.60 Identifying ESA hyporesponsiveness should lead to an intensive search for potentially correctable factors.
The two major factors accounting for the state of hyporesponsiveness are inflammation and iron deficiency.75,76
Inflammation. High C-reactive protein levels have been shown to predict resistance to erythropoietin in dialysis patients.77 The release of cytokines such as tumor necrosis factor alpha, interleukin 1, and interferon gamma has an inhibitory effect on erythropoiesis.78 Additionally, inflammation can alter the response to ESAs by disrupting the metabolism of iron79 through the release of hepcidin, as previously discussed.38 These reasons likely account for the observed lower response to ESAs in the setting of acute illness and explain why ESAs are not recommended for correcting acute anemia.80
Iron deficiency also can blunt the response to ESAs. Large amounts of iron are needed for effective erythropoietic bursts. As such, iron supplementation is now a recognized treatment of renal anemia.81
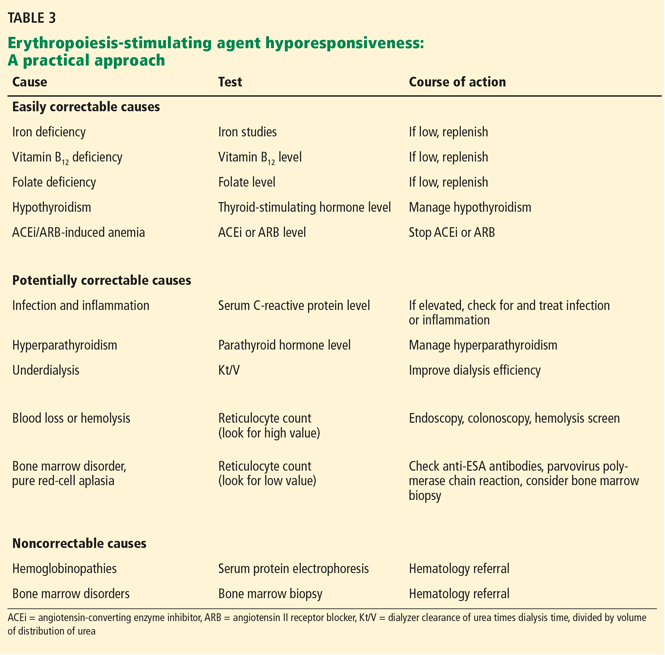
Other factors associated with hyporesponsiveness include chronic occult blood loss, aluminum toxicity, cobalamin or folate deficiencies, testosterone deficiency, inadequate dialysis, hyperparathyroidism, and superimposed primary bone marrow disease,82,83 and these should be addressed in patients whose anemia does not respond as expected to ESA therapy. A summary of the main causes of ESA hyporesponsiveness, their reversibility, and recommended treatments is presented in Table 3.
Antibody-mediated pure red-cell aplasia. Rarely, patients receiving ESA therapy develop antibodies that neutralize both the ESA and endogenous erythropoietin. The resulting syndrome, called antibody-mediated pure red-cell aplasia, is characterized by the sudden development of severe transfusion-dependent anemia. This has historically been connected to epoetin beta, a formulation not in use in the United States. However, cases have been documented with epoetin alfa and darbepoetin. The incidence rate is low with subcutaneous ESA use, estimated at 0.5 cases per 10,000 patient-years84 and anecdotal with intravenous ESA.85 The definitive diagnosis requires demonstration of neutralizing antibodies against erythropoietin. Parvovirus infection should be excluded as an alternative cause of pure redcell aplasia.
ANEMIA IN CANCER PATIENTS
ESAs are effective in raising hemoglobin levels and reducing transfusion requirements in patients with chemotherapy-induced anemia.86 However, there are data linking the use of ESAs to shortened survival in patients who have a variety of solid tumors.87
Several mechanisms have been proposed to explain this rapid disease progression, most notably acceleration in tumor growth88–90 by stimulation of erythropoietin receptors on the surface of the tumor cells, leading to increased tumor angiogenesis.91,92
For these reasons, treatment of renal anemia in the setting of active malignancy should be referred to an oncologist.
NOVEL TREATMENTS
Several new agents for treating renal anemia are currently under review.
Continuous erythropoiesis receptor activator
Continuous erythropoiesis receptor activator is a pegylated form of recombinant human erythropoietin that has the ability to repeatedly activate the erythropoietin receptor. It appears to be similar to the other forms of erythropoietin in terms of safety and efficacy in both end-stage renal disease93 and chronic kidney disease.94 It has the advantage of an extended serum half-life, which allows for longer dosing intervals, ie, every 2 weeks. Its use is currently gaining popularity in the dialysis community.
HIF stabilizers
Our growing understanding of the physiology of erythropoietin offers new potential treatment targets. As previously described, production of erythropoietin is stimulated by HIFs. In order to be degraded, these HIFs are hydroxylated at their proline residues by a prolyl hydroxylase. A new category of drugs called prolyl-hydroxylase inhibitors (PDIs) offers the advantage of stabilizing the HIFs, leading to an increase in erythropoietin production.
In phase 1 and 2 clinical trials, these agents have been shown to increase hemoglobin in both end-stage renal disease and chronic kidney disease patients15,16 but not in anephric patients, demonstrating a renal source of the erythropoietin production even in nonfunctioning kidneys. The study of one PDI agent (FG 2216) was halted temporarily after a report of death from fulminant hepatitis, but the other (FG 4592) continues to be studied in a phase 2 clinical trial.95,96
TAKE-HOME POINTS
- Anemia of renal disease is a common condition that is mainly caused by a decrease in erythropoietin production by the kidneys.
- While anemia of renal disease can be corrected with ESAs, it is necessary to investigate and rule out underlying treatable conditions such as iron or vitamin deficiencies before giving an ESA.
- Anemia of renal disease is associated with significant morbidity such as increased risk of left ventricular hypertrophy, myocardial infarction, and heart failure, and has been described as an all-cause mortality multiplier.
- Unfortunately, the only undisputed benefit of treatment to date remains the avoidance of blood transfusions. Furthermore, the large randomized controlled trials that looked at the benefits of ESA have shown that their use can be associated with increased risk of cardiovascular events. Therefore, use of an ESA in end-stage renal disease should never target a normal hemoglobin levels but rather aim for a hemoglobin level of no more than 11.5 g/dL.
- Use of an ESA in chronic kidney disease should be individualized and is not recommended to be started unless the hemoglobin level is less than 10 g/dL.
- Several newer agents for renal anemia are currently under review. A pegylated form of recombinant human erythropoietin has an extended half-life, and a new and promising category of drugs called HIF stabilizers is currently under study.
- World Health Organization (WHO). Nutritional anaemias: report of a WHO scientific group. Geneva, Switzerland: World Health Organization, 1968.
- Hsu CY, McCulloch CE, Curhan GC, et al. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol 2002; 13:504–510.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoetin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Kazmi WH, Kausz AT, Khan S, et al. Anemia: an early complication of chronic renal insufficiency. Am J Kidney Dis 2001; 38:803–812.
- United States Renal Data System. Chapter 3. Morbidity & mortality in patients with CKD. www.usrds.org/2012/view/v1_03.aspx. Accessed June 9, 2016.
- Horwich TB, Fonarow GC, Hamilton MA, MacLellan WR, Borenstein J. Anemia is associated with worse symptoms, greater impairment in functional capacity and a significant increase in mortality in patients with advanced heart failure. J Am Coll Cardiol 2002; 39:1780–1786.
- Mark DB, Felker GM. B-type natriuretic peptide: a biomarker for all seasons? N Engl J Med 2004; 350:718–720.
- Walker AM, Schneider G, Yeaw J, Nordstrom B, Robbins S, Pettitt D. Anemia as a predictor of cardiovascular events in patients with elevated serum creatinine. J Am Soc Nephrol 2006; 17:2293–2298.
- Abramson JL, Jurkovitz CT, Vaccarino V, Weintraub WS, McClellan W. Chronic kidney disease, anemia, and incident stroke in a middle-aged, community-based population: the ARIC Study. Kidney Int 2003; 64:610–615.
- Sarnak MJ, Tighiouart H, Manjunath G, et al. Anemia as a risk factor for cardiovascular disease in the Atherosclerosis Risk in Communities (ARIC) study. J Am Coll Cardiol 2002; 40:27–33.
- McClellan WM, Flanders WD, Langston RD, Jurkovitz C, Presley R. Anemia and renal insufficiency are independent risk factors for death among patients with congestive heart failure admitted to community hospitals: a population-based study. J Am Soc Nephrol 2002; 13:1928–1936.
- Xia H, Ebben J, Ma JZ, Collins AJ. Hematocrit levels and hospitalization risks in hemodialysis patients. J Am Soc Nephrol 1999; 10:1309–1316.
- Collins AJ, Li S, St Peter W, et al. Death, hospitalization, and economic associations among incident hemodialysis patients with hematocrit values of 36 to 39%. J Am Soc Nephrol 2001; 12:2465–2473.
- Agarwal AK. Practical approach to the diagnosis and treatment of anemia associated with CKD in elderly. J Am Med Dir Assoc 2006; 7(suppl 9):S7–S12.
- Bernhardt WM, Wiesener MS, Scigalla P, et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol 2010; 21:2151–2156.
- Provenzano R, Fadda G, Bernardo M, et al. FG-2216, a novel oral HIF-PHI, stimulates erythropoiesis and increases hemoglobin concentration in patients with non-dialysis CKD. Am J Kidney Dis 2008; 51:B80.
- Maxwell PH, Osmond MK, Pugh CW, et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int 1993; 44:1149–1162.
- Maxwell PH, Ferguson DJ, Nicholls LG, et al. Sites of erythropoietin production. Kidney Int 1997; 51:393–401.
- Jelkmann W. Erythropoeitin: structure, control of production and function. Physiol Rev 1992; 72:449–489.
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92:5510–5514.
- Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 1995; 270:1230–1237.
- Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399:271–275.
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 1997; 272:22642–22647.
- Malik J, Kim AR, Tyre KA, Cherukuri AR, Palis J. Erythropoietin critically regulates the terminal maturation of murine and human primitive erythroblasts. Haematologica 2013; 98:1778–1787.
- Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 1995; 83:59–67.
- Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(-/-)5b(-/-) mice due to decreased survival of early erythroblasts. Blood 2001; 98:3261–3273.
- Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol 2005; 202:199–211.
- Conrad ME, Umbreit JN. Pathways of iron absorption. Blood Cells Mol Dis 2002; 29:336–355.
- Frazer DM, Anderson GJ. The orchestration of body iron intake: how and where do enterocytes receive their cues? Blood Cells Moll Dis 2003; 30:288–297.
- Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 2005; 1:191–200.
- Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999; 21:195–199.
- Bothwell TH. Overview and mechanisms of iron regulation. Nutr Rev 1995: 53:237–245.
- Kawabata H, Nakamaki T, Ikonomi P, Smith RD, Germain RS, Koeffler HP. Expression of transferrin receptor 2 in normal and neoplastic hematopoietic cells. Blood 2001; 98:2714–2719.
- Arosio P, Levi S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim Biophys Acta 2010; 1800:783–792.
- Finch CA, Bellotti V, Stray S, et al. Plasma ferritin determination as a diagnostic tool. West J Med 1986; 145:657–663.
- Delaby C, Pilard N, Goncalves AS, Beaumont C, Canonne-Hergaux F. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 2005; 106:3979–3984.
- Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003; 102:783–788.
- Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002; 110:1037–1044.
- DeGowin RL, Lavender AR, Forland M, Charleston D, Gottschalk A. Erythropoiesis and erythropoietin in patients with chronic renal failure treated with hemodialysis and testosterone. Ann Intern Med 1970; 72:913–918.
- Richardson JR Jr, Weinstein MB. Erythropoietic response of dialyzed patients to testosterone administration. Ann Intern Med 1970; 73:403–407
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Intern Med 1989; 111:992–1000.
- Moreno F, Aracil FJ, Pérez R, Valderrábano F. Controlled study on the improvement of quality of life in elderly hemodialysis patients after correcting end-stage renal disease-related anemia with erythropoietin. Am J Kidney Dis 1996; 27:548–556.
- Nissenson AR, Nimer SD, Wolcott DL. Recombinant human erythropoietin and renal anemia: molecular biology, clinical efficacy, and nervous system effects. Ann Intern Med 1991; 114:402–416.
- Stivelman JC. Benefits of anaemia treatment on cognitive function. Nephrol Dial Transplant 2000; 15(suppl 3):29–35.
- Maddux FW, Shetty S, del Aguila MA, Nelson MA, Murray BM. Effect of erythropoiesis-stimulating agents on healthcare utilization, costs, and outcomes in chronic kidney disease. Ann Pharmacother 2007; 41:1761–1769.
- Macdougall IC, Lewis NP, Saunders MJ, et al. Long-term cardiorespiratory effects of amelioration of renal anaemia by erythropoietin. Lancet 1990; 335:489–493.
- Silverberg DS, Wexler D, Blum M, et al. Effects of treatment with epoetin beta on outcomes in patients with anaemia and chronic heart failure. Kidney Blood Press Res 2005; 28:41–47.
- Perkins R, Olson S, Hansen J, Lee J, Stiles K, Lebrun C. Impact of an anemia clinic on emergency room visits and hospitalizations in patients with anemia of CKD pre-dialysis. Nephrol Nurs J 2007; 34:167–173, 182.
- Locatelli F, Conte F, Marcelli D. The impact of haematocrit levels and erythropoietin treatment on overall and cardiovascular mortality and morbidity—the experience of the Lombardy Dialysis Registry. Nephrol Dial Transplant 1998; 13:1642–1644.
- Centers for Medicare and Medicaid Services; Kinney R. 2005 Annual Report: ESRD Clinical Performance Measures Project. Am J Kidney Dis 2006; 48(suppl 2):S1–S106.
- US Renal Data System. Annual Data Report 2006. www.usrds.org/adr.aspx. Accessed July 3, 2016.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590.
- Singh AK, Szczech L, Tang KL, et al; CHOIR Investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- Pfeffer MA, Burdmann EA, Chen CY, et al; TREAT Investigators. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 2009; 361:2019–2032.
- Kirkpantur A, Kahraman S, Yilmaz R, et al. The effects of maintenance recombinant human erythropoietin therapy on ambulatory blood pressure recordings: conventional, Doppler, and tissue Doppler echocardiographic parameters. Artif Organs 2005; 29:965–972.
- Fishbane S, Berns JS. Hemoglobin cycling in hemodialysis patients treated with recombinant human erythropoietin. Kidney Int 2005; 68:1337–1343.
- Szczech LA, Barnhart HX, Inrig JK, et al. Secondary analysis of the CHOIR trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int 2008; 74:791–798.
- Solomon SD, Uno H, Lewis EF, et al; Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) Investigators. Erythropoietic response and outcomes in kidney disease and type 2 diabetes. N Engl J Med 2010; 363:1146–1155.
- Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney Int Suppl 2012; 2:279–335.
- Fernández-Rodríguez AM, Guindeo-Casasús MC, Molero-Labarta T, et al. Diagnosis of iron deficiency in chronic renal failure. Am J Kidney Dis 1999; 34:508–513.
- Eschbach JW, Cook JD, Scribner BH, Finch CA. Iron balance in hemodialysis patients. Ann Intern Med 1977; 87:710–713.
- Mittman N, Sreedhara R, Mushnick R, et al. Reticulocyte hemoglobin content predicts functional iron deficiency in hemodialysis patients receiving rHuEPO. Am J Kidney Dis 1997; 30:912–922.
- Tessitore N, Solero GP, Lippi G, et al. The role of iron status markers in predicting response to intravenous iron in haemodialysis patients on maintenance erythropoietin. Nephrol Dial Transplant 2001; 16:1416–1423.
- Coyne DW. Iron indices: what do they really mean? Kidney Int Suppl 2006; 101:S4–S8.
- Fishbane S, Kowalski EA, Imbriano LJ, Maesaka JK. The evaluation of iron status in hemodialysis patients. J Am Soc Nephrol 1996; 7:2654–2657.
- Coyne DW, Kapoian T, Suki W, et al; DRIVE Study Group. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: results of the Dialysis Patients’ Response to IV Iron with Elevated Ferritin (DRIVE) Study. J Am Soc Nephrol 2007; 18:975–984.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoietin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Korte W, Cogliatti SB, Jung K, Riesen W. Mild renal dysfunction is sufficient to induce erythropoietin deficiency in patients with unexplained anaemia. Clin Chim Acta 2000; 292:149–154.
- Locatelli F, Olivares J, Walker R, et al; European/Australian NESP 980202 Study Group. Novel erythropoiesis stimulating protein for treatment of anemia in chronic renal insufficiency. Kidney Int 2001; 60:741–747.
- Carrera F, Burnier M. Use of darbepoetin alfa in the treatment of anaemia of chronic kidney disease: clinical and pharmacoeconomic considerations. NDT Plus 2009; 2(suppl 1):i9–i17.
- Egrie JC, Browne JK. Development and characterization of novel erythropoiesis stimulating protein (NESP). Nephrol Dial Transplant 2001; 16(suppl 3):3–13.
- Nissenson AR, Charytan C. Controversies in iron management. Kidney Int Suppl 2003; 87:S64–S71.
- Kilpatrick RD, Critchlow CW, Fishbane S, et al. Greater epoetin alpha responsiveness is associated with improved survival in hemodialysis patients. Clin J Am Soc Nephrol 2008; 3:1077–1083.
- Locatelli F, Aljama P, Barany P, et al; European Best Practice Guidelines Working Group. Revised European best practice guidelines for the management of anaemia in patients with chronic renal failure. Nephrol Dial Transplant 2004; 19(suppl 2):ii1–ii47.
- Stenvinkel P. The role of inflammation in the anaemia of end-stage renal disease. Nephrol Dial Transplant 2001; 16(suppl 7):36–40.
- Barany P, Divino Filho JC, Bergstrom J. High C-reactive protein is a strong predictor of resistance to erythropoietin in hemodialysis patients. Am J Kidney Dis 1997; 29:565–568.
- Drueke T. Hyporesponsiveness to recombinant human erythropoietin. Nephrol Dial Transplant 2001; 16(suppl 7):25–28.
- Casadevall N. Cellular mechanism of resistance to erythropoietin. Nephrol Dial Transplant 1995; 10(suppl 6):27–30.
- Kraus E, Rabb H. EPO therapy during acute kidney disease: to use or not to use, that is the question. Am J Kidney Dis 2005; 46:967–969.
- Gotloib L, Silverberg D, Fudin R, Shostak A. Iron deficiency is a common cause of anemia in chronic kidney disease and can often be corrected with intravenous iron. J Nephrol 2006; 19:161–167.
- Tarng DC, Huang TP, Chen TW, Yang WC. Erythropoietin hyporesponsiveness: from iron deficiency to iron overload. Kidney Int Suppl 1999; 69:S107–S118.
- Drüeke TB. Modulating factors in the hematopoietic response to erythropoietin. Am J Kidney Dis 1991; 18(suppl 1):87–92.
- Boven K, Stryker S, Knight J, et al. The increased incidence of pure red cell aplasia with an Eprex formulation in uncoated rubber stopper syringes. Kidney Int 2005; 67:2346–2353.
- Shimizu H, Saitoh T, Ota F, et al. Pure red cell aplasia induced only by intravenous administration of recombinant human erythropoietin. Acta Haematol 2011; 126:114–118.
- Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst Rev 2012; 12:CD003407.
- Bohlius J, Langensiepen S, Schwarzer G, et al. Recombinant human erythropoietin and overall survival in cancer patients: results of a comprehensive meta-analysis. J Natl Cancer Inst 2005; 97:489–498.
- Henke M, Laszig R, Rübe C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet 2003; 362:1255–1260.
- Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005; 23:5960–5972.
- Brower V. Erythropoietin may impair, not improve, cancer survival. Nat Med 2003; 9:1439.
- Acs G, Acs P, Beckwith SM, et al. Erythropoietin and erythropoietin receptor expression in human cancer. Cancer Res 2001; 61:3561–3565.
- Yasuda Y, Fujita Y, Matsuo T, et al. Erythropoietin regulates tumour growth of human malignancies. Carcinogenesis 2003; 24:1021–1029.
- Levin NW, Fishbane S, Cañedo FV, et al; MAXIMA Study Investigators. Intravenous methoxy polyethylene glycol-epoetin beta for haemoglobin control in patients with chronic kidney disease who are on dialysis: a randomised non-inferiority trial (MAXIMA). Lancet 2007; 370:1415–1421.
- Macdougall IC, Walker R, Provenzano R, et al; ARCTOS Study Investigators. C.E.R.A. corrects anemia in patients with chronic kidney disease not on dialysis: results of a randomized clinical trial. Clin J Am Soc Nephrol 2008; 3:337–347.
- Frohna PA, Milwee S, Pinkett J, et al. Preliminary results from a randomized, single-blind, placebo-controlled trial of FG-4592, a novel hypoxia inducible factor prolyl hydroxylase inhibitor, in subjects with CKD anemia (abstract). J Am Soc Nephrol 2007; 18:763.
- Holdstock L, Meadowcroft AM, Maier R, et al. Four-week studies of oral hypoxia-inducible factor-prolyl hydroxylase inhibitor GSK1278863 for treatment of anemia. J Am Soc Nephrol 2016; 27:1234–1244.
Anemia is a frequent complication of chronic kidney disease, occurring in over 90% of patients receiving renal replacement therapy. It is associated with significant morbidity and mortality. While its pathogenesis is typically multifactorial, the predominant cause is failure of the kidneys to produce enough endogenous erythropoietin. The clinical approval of recombinant human erythropoietin in 1989 dramatically changed the treatment of anemia of chronic kidney disease, but randomized controlled trials yielded disappointing results when erythropoiesis-stimulating agents (ESAs) were used to raise hemoglobin to normal levels.
This article reviews the epidemiology and pathophysiology of anemia of chronic kidney disease and discusses the complicated and conflicting evidence regarding its treatment.
DEFINITION AND PREVALENCE
Anemia is defined as a hemoglobin concentration less than 13.0 g/dL for men and less than 12.0 g/dL for premenopausal women.1 It is more common in patients with impaired kidney function, especially when the glomerular filtration rate (GFR) falls below 60 mL/min. It is rare at GFRs higher than 80 mL/min,2 but as the GFR falls, the severity of the anemia worsens3 and its prevalence increases: almost 90% of patients with a GFR less than 30 mL/min are anemic.4
RENAL ANEMIA IS ASSOCIATED WITH BAD OUTCOMES
Anemia in chronic kidney disease is independently associated with risk of death. It is also an all-cause mortality multiplier, ie, it magnifies the risk of death from other disease states.5
In observational studies, anemia was associated with faster progression of left ventricular hypertrophy, inflammation, and increased myocardial and peripheral oxygen demand, thereby leading to worse cardiac outcomes with increased risk of myocardial infarction, coronary revascularization, and readmission for heart failure.6–8 Anemia is also associated with fatigue, depression, reduced exercise tolerance, stroke, and increased risk of rehospitalization.9–13
RENAL ANEMIA IS MULTIFACTORIAL
Anemia of chronic kidney disease is typically attributed to the decrease of erythropoietin production that accompanies the fall in GFR. However, the process is multifactorial, with several other contributing factors: absolute and functional iron deficiency, folate and vitamin B12 deficiencies, reduced red blood cell life span, and suppression of erythropoiesis by the uremic milieu.14
While it was once thought that chronic kidney disease leads to loss of erythropoietin-producing cells, it is now known that downregulation of hypoxia-inducible factor (HIF; a transcription factor) is at least partially responsible for the decrease in erythropoietin production15,16 and that this downregulation is reversible (see below).
ERYTHROPOIETIN, IRON, AND RED BLOOD CELLS
Erythropoietin production is triggered by hypoxia, mediated by HIF
Erythropoietin is produced primarily in the deep cortex and outer medulla of the kidneys by a special population of peritubular interstitial cells.17 The parenchymal cells of the liver also produce erythropoietin, but much less.18
The rate of renal erythropoietin synthesis is determined by tissue oxygenation rather than by renal blood flow; production increases as the hemoglobin concentration drops and the arterial oxygen tension decreases (Figure 1).19
The gene for erythropoietin is located on chromosome 7 and is regulated by HIF. HIF molecules are composed of an alpha subunit, which is unstable at high Po2, and a beta subunit, constitutively present in the nucleus.20
In hypoxic conditions, the HIF dimer is transcriptionally active and binds to specific DNA recognition sequences called hypoxia-response elements. Gene transcription is upregulated, leading to increased production of erythropoietin.21
Under normal oxygen tension, on the other hand, the proline residue of the HIF alpha subunit is hydroxylated. The hydroxylated HIF alpha subunit is then degraded by proteasomal ubiquitylation, which is mediated by the von Hippel-Lindau tumor-suppressor gene pVHL.22 Degradation of HIF alpha prevents formation of the HIF heterodimers. HIF therefore cannot bind to the hypoxia-response elements, and erythropoietin gene transcription does not occur.23
Thus, in states of hypoxia, erythropoietin production is upregulated, whereas with normal oxygen tension, production is downregulated.
Erythropoietin is essential for terminal maturation of erythrocytes
Erythropoietin is essential for terminal maturation of erythrocytes.24 It is thought to stimulate the growth of erythrogenic progenitors: burst-forming units-erythroid (BFU-E) and colony-forming units-erythroid (CFU-E). In the absence of erythropoietin, BFU-E and CFU-E fail to differentiate into mature erythrocytes.25
Binding of erythropoietin to its receptor sets off a series of downstream signals, the most important being the signal transducer and activator of transcription 5 (STAT5). In animal studies, STAT5 was found to inhibit apoptosis through the early induction of an antiapoptotic gene, Bcl-xL.26
Iron metabolism is controlled by several proteins
Iron is characterized by its capacity to accept or donate electrons. This unique property makes it a crucial element in many biochemical reactions such as enzymatic activity, DNA synthesis, oxygen transport, and cell respiration.
Iron metabolism is under the control of several proteins that play different roles in its absorption, recycling, and loss (Figure 2).27
Dietary iron exists primarily in its poorly soluble trivalent ferric form (Fe3+), and it needs to be reduced to its soluble divalent ferrous form (Fe2+) by ferric reductase to be absorbed. Ferrous iron is taken up at the apical side of enterocytes by a divalent metal transporter (DMT1) and is transported across the brush border.28
To enter the circulation, iron has to be transported across the basolateral membrane by a transporter called ferroportin.29 Ferroportin is also found in placental syncitiotrophoblasts, where it transfers iron from mother to fetus, and in macrophages, where it allows recycling of iron scavenged from damaged cells back into the circulation.30 Upon its release, the ferrous iron is oxidized to the ferric form and loaded onto transferrin. This oxidation process involves hephaestin, a homologue of the ferroxidase ceruloplasmin.31
In the plasma, iron is bound to transferrin, and under normal circumstances one-third of transferrin is saturated with iron.32 Transferrin receptors are present on most cells but are most dense on erythroid precursors. Each transferrin receptor can bind two transferrin molecules. After binding to transferrin, the transferrin receptor is endocytosed, and the iron is released into acidified vacuoles. The transferrin-receptor complex is then recycled to the surface.33
Ferritin is the cellular storage protein for iron, and it can store up to 4,500 atoms of iron within its spherical cavity.34 The serum level of ferritin reflects overall storage, with 1 ng/mL of ferritin indicating 10 mg of total iron stores.35 Ferritin is also an acute-phase reactant, and plasma levels can increase in inflammatory states such as infection or malignancy. As such, elevated ferritin does not necessarily indicate elevated iron stores.
Iron is lost in sweat, shed skin cells, and sloughed intestinal mucosal cells. However, there is no specific mechanism of iron excretion from the human body. Thus, iron is mainly regulated at the level of intestinal absorption. The iron exporter ferroportin is upregulated by the amount of available iron and is degraded by hepcidin.36
Hepcidin is a small cysteine-rich cationic peptide that is primarily produced in the liver, with some minor production also occurring in the kidneys.37 Transcription of the gene encoding hepcidin is downregulated by anemia and hypoxia and upregulated by inflammation and elevated iron levels.38 Transcription of hepcidin leads to degradation of ferroportin and a decrease in intestinal iron absorption. On the other hand, anemia and hypoxia inhibit hepcidin transcription, which allows ferroportin to facilitate intestinal iron absorption.
TREATMENT OF RENAL ANEMIA
Early enthusiasm for erythropoietin agents
Androgens started to be used to treat anemia of end-stage renal disease in 1970,39,40 and before the advent of recombinant human erythropoietin, they were a mainstay of nontransfusional therapy for anemic patients on dialysis.
The approval of recombinant human erythropoietin in 1989 drastically shifted the treatment of renal anemia. While the initial goal of treating anemia of chronic kidney disease with erythropoietin was to prevent blood transfusions,41 subsequent studies showed that the benefits might be far greater. Indeed, an initial observational trial showed that erythropoiesis-stimulating agents (ESAs) were associated with improved quality of life,42 improved neurocognitive function,43,44 and even cost savings.45 The benefits also extended to major outcomes such as regression of left ventricular hypertrophy,46 improvement in New York Heart Association class and cardiac function,47 fewer hospitalizations,48 and even reduction of cardiovascular mortality rates.49
As a result, ESA use gained popularity, and by 2006 an estimated 90% of dialysis patients were receiving these agents.50 The target and achieved hemoglobin levels also increased, with mean hemoglobin levels in hemodialysis patients being raised from 9.7 to 12 g/dL.51
Disappointing results in clinical trials of ESAs to normalize hemoglobin
To prospectively study the effects of normalized hemoglobin targets, four randomized controlled trials were conducted (Table 1):
- The Normal Hematocrit Study (NHCT)52
- The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial53
- The Cardiovascular Risk Reduction by Early Anemia Treatment (CREATE) trial54
- The Trial to Reduce Cardiovascular Events With Aranesp Therapy (TREAT).55
These trials randomized patients to either higher “normal-range” hemoglobin targets or to lower target hemoglobin levels.
Their findings were disappointing and raised several red flags about excessive use of ESAs. The trials found no benefit in higher hemoglobin targets, and in fact, some of them demonstrated harm in patients randomized to higher targets. Notably, higher hemoglobin targets were associated with significant side effects such as access-site thrombosis,52 strokes,55 and possibly cardiovascular events.54,55 Only the CREATE trial was able to demonstrate a quality-of-life benefit for the high-target group.54
It remains unclear whether these adverse events were from the therapy itself or from an increased morbidity burden in the treated patients. Erythropoietin use is associated with hypertension,56 thought to be related to endothelin-mediated vasoconstriction.57 In our experience, this is most evident when hemoglobin levels are normalized with ESA therapy. Cycling of erythropoietin levels between extreme levels can lead to vascular remodeling, which may also be related to its cardiovascular effects.57
A noticeable finding in several of these trials was that patients failed to achieve the higher hemoglobin target despite the use of very high doses of ESA. Reanalysis of data from the CHOIR and CREATE trials showed that the patients who had worse outcomes were more likely to have required very high doses without achieving their target hemoglobin.58,59 Indeed, patients who achieved the higher target hemoglobin levels, usually at lower ESA doses, had better outcomes. This suggested that the need for a higher dose was associated with poorer outcomes, either as a marker of comorbidity or due to yet undocumented side effects of such high doses.
General approach to therapy
Before attributing anemia to chronic kidney disease, a thorough evaluation should be conducted to look for any reversible process that could be contributing to the anemia.
The causes of anemia are numerous and beyond the scope of this review. However, among the common causes of anemia in chronic kidney disease are deficiencies of iron, vitamin B12, and folate. Therefore, guidelines recommend checking iron, vitamin B12, and folate levels in the initial evaluation of anemia.60
Iron deficiency in particular is very common in chronic kidney disease patients and is present in nearly all dialysis patients.61 Hemodialysis patients are estimated to lose 1 to 3 g of iron per year as a result of blood loss in the dialysis circuit and increased iron utilization secondary to ESA therapy.62
However, in contrast to the general population, in which the upper limits of normal for iron indices are well defined, high serum ferritin levels appear to be poorly predictive of hemoglobin responsiveness in dialysis patients.63,64 Thus, the cutoffs that define iron responsiveness are much higher than standard definitions for iron deficiency.65,66 The Dialysis Patients’ Response to IV Iron With Elevated Ferritin (DRIVE) study showed that dialysis patients benefit from intravenous iron therapy even if their ferritin is as high as 1,200 ng/mL, provided their transferrin saturation is below 30%.67
Of note, erythropoietin levels cannot be used to distinguish renal anemia from other causes of anemia. Indeed, patients with renal failure may have “relative erythropoietin deficiency,” ie, “normal” erythropoietin levels that are actually too low in view of the degree of anemia.68,69 In addition to the decreased production capacity by the kidney, there appears to be a component of resistance to the action of erythropoietin in the bone marrow.
For these reasons, there is no erythropoietin level that can be considered “inadequate” or defining of renal anemia. Thus, measuring erythropoietin levels is not routinely recommended in the evaluation of renal anemia.
Two ESA preparations
The two ESAs that have traditionally been used in the treatment of renal anemia are recombinant human erythropoietin and darbepoietin alfa. They appear to be equivalent in terms of safety and efficacy.70 However, darbepoietin alfa has more sialic acid molecules, giving it a higher potency and longer half-life and allowing for less-frequent injections.71,72
In nondialysis patients, recombinant human erythropoietin is typically given every 1 to 2 weeks, whereas darbepoietin alfa can be given every 2 to 4 weeks. In dialysis patients, recombinant human erythropoietin is typically given 3 times per week with every dialysis treatment, while darbepoietin alfa is given once a week.
Target hemoglobin levels: ≤ 11.5 g/dL
In light of the four trials described in Table 1, the international Kidney Disease: Improving Global Outcomes (KDIGO) guidelines60 recommend the following (Table 2):
For patients with chronic kidney disease who are not on dialysis, ESA therapy should not be initiated if the hemoglobin level is higher than 10 g/dL. If the hemoglobin level is lower than 10 g/dL, ESA therapy can be initiated, but the decision needs to be individualized based on the rate of fall of hemoglobin concentration, prior response to iron therapy, the risk of needing a transfusion, the risks related to ESA therapy, and the presence of symptoms attributable to anemia.
For patients on dialysis, ESA therapy should be used when the hemoglobin level is between 9 and 10 g/dL to avoid having the hemoglobin fall below 9 g/dL.
In all adult patients, ESAs should not be used to intentionally increase the hemoglobin level above 13 g/dL but rather to maintain levels no higher than 11.5 g/dL. This target is based on the observation that adverse outcomes were associated with ESA use with hemoglobin targets higher than 13 g/dL (Table 1).
Target iron levels
Regarding iron stores, the guidelines recommend the following:
For adult patients with chronic kidney disease who are not on dialysis, iron should be given to keep transferrin saturation above 20% and ferritin above 100 ng/mL. Transferrin saturation should not exceed 30%, and ferritin levels should not exceed 500 ng/mL.
For adult patients on dialysis, iron should be given to maintain transferrin saturation above 30% and ferritin above 200 ng/mL.
The upper limits of ferritin and transferrin saturation are somewhat controversial, as the safety of intentionally maintaining respective levels greater than 30% and 500 ng/mL has been studied in very few patients. Transferrin saturation should in general not exceed 50%.
High ferritin levels are associated with higher death rates, but whether elevation of ferritin levels is a marker of excessive iron administration rather than a nonspecific acute-phase reactant is not clear. The 2006 guidelines60 cited upper ferritin limits of 500 to 800 ng/mL. However, the more recent DRIVE trial67 showed that patients with ferritin levels of 500 to 1,200 ng/mL will respond to intravenous administration of iron with an increase in their hemoglobin levels. This has led many clinicians to adopt a higher ferritin limit of 1,200 ng/mL.
Hemosiderosis, or excess iron deposition, was a known consequence of frequent transfusions in patients with end-stage renal disease before ESA therapy was available. However, there have been no documented cases of clinical iron overload from iron therapy using current guidelines.73
These algorithms are nuanced, and the benefit of giving intravenous iron should always be weighed against the risks of short-term acute toxicity and infection. Treatment of renal anemia not only requires in-depth knowledge of the topic, but also familiarity with the patient’s specific situation. As such, it is not recommended that clinicians unfamiliar with the treatment of renal anemia manage its treatment.
PARTICULAR CIRCUMSTANCES
Inflammation and ESA resistance
While ESAs are effective in treating anemia in many cases, in many patients the anemia fails to respond. This is of particular importance, since ESA hyporesponsiveness has been found to be a powerful predictor of cardiovascular events and death.74 It is unclear, however, whether high doses of ESA are inherently toxic or whether hyporesponsiveness is a marker of adverse outcomes related to comorbidities.
KDIGO defines initial hyporesponsiveness as having no increase in hemoglobin concentration after the first month of appropriate weight-based dosing, and acquired hyporesponsiveness as requiring two increases in ESA doses up to 50% beyond the dose at which the patient had originally been stable.60 Identifying ESA hyporesponsiveness should lead to an intensive search for potentially correctable factors.
The two major factors accounting for the state of hyporesponsiveness are inflammation and iron deficiency.75,76
Inflammation. High C-reactive protein levels have been shown to predict resistance to erythropoietin in dialysis patients.77 The release of cytokines such as tumor necrosis factor alpha, interleukin 1, and interferon gamma has an inhibitory effect on erythropoiesis.78 Additionally, inflammation can alter the response to ESAs by disrupting the metabolism of iron79 through the release of hepcidin, as previously discussed.38 These reasons likely account for the observed lower response to ESAs in the setting of acute illness and explain why ESAs are not recommended for correcting acute anemia.80
Iron deficiency also can blunt the response to ESAs. Large amounts of iron are needed for effective erythropoietic bursts. As such, iron supplementation is now a recognized treatment of renal anemia.81
Other factors associated with hyporesponsiveness include chronic occult blood loss, aluminum toxicity, cobalamin or folate deficiencies, testosterone deficiency, inadequate dialysis, hyperparathyroidism, and superimposed primary bone marrow disease,82,83 and these should be addressed in patients whose anemia does not respond as expected to ESA therapy. A summary of the main causes of ESA hyporesponsiveness, their reversibility, and recommended treatments is presented in Table 3.
Antibody-mediated pure red-cell aplasia. Rarely, patients receiving ESA therapy develop antibodies that neutralize both the ESA and endogenous erythropoietin. The resulting syndrome, called antibody-mediated pure red-cell aplasia, is characterized by the sudden development of severe transfusion-dependent anemia. This has historically been connected to epoetin beta, a formulation not in use in the United States. However, cases have been documented with epoetin alfa and darbepoetin. The incidence rate is low with subcutaneous ESA use, estimated at 0.5 cases per 10,000 patient-years84 and anecdotal with intravenous ESA.85 The definitive diagnosis requires demonstration of neutralizing antibodies against erythropoietin. Parvovirus infection should be excluded as an alternative cause of pure redcell aplasia.
ANEMIA IN CANCER PATIENTS
ESAs are effective in raising hemoglobin levels and reducing transfusion requirements in patients with chemotherapy-induced anemia.86 However, there are data linking the use of ESAs to shortened survival in patients who have a variety of solid tumors.87
Several mechanisms have been proposed to explain this rapid disease progression, most notably acceleration in tumor growth88–90 by stimulation of erythropoietin receptors on the surface of the tumor cells, leading to increased tumor angiogenesis.91,92
For these reasons, treatment of renal anemia in the setting of active malignancy should be referred to an oncologist.
NOVEL TREATMENTS
Several new agents for treating renal anemia are currently under review.
Continuous erythropoiesis receptor activator
Continuous erythropoiesis receptor activator is a pegylated form of recombinant human erythropoietin that has the ability to repeatedly activate the erythropoietin receptor. It appears to be similar to the other forms of erythropoietin in terms of safety and efficacy in both end-stage renal disease93 and chronic kidney disease.94 It has the advantage of an extended serum half-life, which allows for longer dosing intervals, ie, every 2 weeks. Its use is currently gaining popularity in the dialysis community.
HIF stabilizers
Our growing understanding of the physiology of erythropoietin offers new potential treatment targets. As previously described, production of erythropoietin is stimulated by HIFs. In order to be degraded, these HIFs are hydroxylated at their proline residues by a prolyl hydroxylase. A new category of drugs called prolyl-hydroxylase inhibitors (PDIs) offers the advantage of stabilizing the HIFs, leading to an increase in erythropoietin production.
In phase 1 and 2 clinical trials, these agents have been shown to increase hemoglobin in both end-stage renal disease and chronic kidney disease patients15,16 but not in anephric patients, demonstrating a renal source of the erythropoietin production even in nonfunctioning kidneys. The study of one PDI agent (FG 2216) was halted temporarily after a report of death from fulminant hepatitis, but the other (FG 4592) continues to be studied in a phase 2 clinical trial.95,96
TAKE-HOME POINTS
- Anemia of renal disease is a common condition that is mainly caused by a decrease in erythropoietin production by the kidneys.
- While anemia of renal disease can be corrected with ESAs, it is necessary to investigate and rule out underlying treatable conditions such as iron or vitamin deficiencies before giving an ESA.
- Anemia of renal disease is associated with significant morbidity such as increased risk of left ventricular hypertrophy, myocardial infarction, and heart failure, and has been described as an all-cause mortality multiplier.
- Unfortunately, the only undisputed benefit of treatment to date remains the avoidance of blood transfusions. Furthermore, the large randomized controlled trials that looked at the benefits of ESA have shown that their use can be associated with increased risk of cardiovascular events. Therefore, use of an ESA in end-stage renal disease should never target a normal hemoglobin levels but rather aim for a hemoglobin level of no more than 11.5 g/dL.
- Use of an ESA in chronic kidney disease should be individualized and is not recommended to be started unless the hemoglobin level is less than 10 g/dL.
- Several newer agents for renal anemia are currently under review. A pegylated form of recombinant human erythropoietin has an extended half-life, and a new and promising category of drugs called HIF stabilizers is currently under study.
Anemia is a frequent complication of chronic kidney disease, occurring in over 90% of patients receiving renal replacement therapy. It is associated with significant morbidity and mortality. While its pathogenesis is typically multifactorial, the predominant cause is failure of the kidneys to produce enough endogenous erythropoietin. The clinical approval of recombinant human erythropoietin in 1989 dramatically changed the treatment of anemia of chronic kidney disease, but randomized controlled trials yielded disappointing results when erythropoiesis-stimulating agents (ESAs) were used to raise hemoglobin to normal levels.
This article reviews the epidemiology and pathophysiology of anemia of chronic kidney disease and discusses the complicated and conflicting evidence regarding its treatment.
DEFINITION AND PREVALENCE
Anemia is defined as a hemoglobin concentration less than 13.0 g/dL for men and less than 12.0 g/dL for premenopausal women.1 It is more common in patients with impaired kidney function, especially when the glomerular filtration rate (GFR) falls below 60 mL/min. It is rare at GFRs higher than 80 mL/min,2 but as the GFR falls, the severity of the anemia worsens3 and its prevalence increases: almost 90% of patients with a GFR less than 30 mL/min are anemic.4
RENAL ANEMIA IS ASSOCIATED WITH BAD OUTCOMES
Anemia in chronic kidney disease is independently associated with risk of death. It is also an all-cause mortality multiplier, ie, it magnifies the risk of death from other disease states.5
In observational studies, anemia was associated with faster progression of left ventricular hypertrophy, inflammation, and increased myocardial and peripheral oxygen demand, thereby leading to worse cardiac outcomes with increased risk of myocardial infarction, coronary revascularization, and readmission for heart failure.6–8 Anemia is also associated with fatigue, depression, reduced exercise tolerance, stroke, and increased risk of rehospitalization.9–13
RENAL ANEMIA IS MULTIFACTORIAL
Anemia of chronic kidney disease is typically attributed to the decrease of erythropoietin production that accompanies the fall in GFR. However, the process is multifactorial, with several other contributing factors: absolute and functional iron deficiency, folate and vitamin B12 deficiencies, reduced red blood cell life span, and suppression of erythropoiesis by the uremic milieu.14
While it was once thought that chronic kidney disease leads to loss of erythropoietin-producing cells, it is now known that downregulation of hypoxia-inducible factor (HIF; a transcription factor) is at least partially responsible for the decrease in erythropoietin production15,16 and that this downregulation is reversible (see below).
ERYTHROPOIETIN, IRON, AND RED BLOOD CELLS
Erythropoietin production is triggered by hypoxia, mediated by HIF
Erythropoietin is produced primarily in the deep cortex and outer medulla of the kidneys by a special population of peritubular interstitial cells.17 The parenchymal cells of the liver also produce erythropoietin, but much less.18
The rate of renal erythropoietin synthesis is determined by tissue oxygenation rather than by renal blood flow; production increases as the hemoglobin concentration drops and the arterial oxygen tension decreases (Figure 1).19
The gene for erythropoietin is located on chromosome 7 and is regulated by HIF. HIF molecules are composed of an alpha subunit, which is unstable at high Po2, and a beta subunit, constitutively present in the nucleus.20
In hypoxic conditions, the HIF dimer is transcriptionally active and binds to specific DNA recognition sequences called hypoxia-response elements. Gene transcription is upregulated, leading to increased production of erythropoietin.21
Under normal oxygen tension, on the other hand, the proline residue of the HIF alpha subunit is hydroxylated. The hydroxylated HIF alpha subunit is then degraded by proteasomal ubiquitylation, which is mediated by the von Hippel-Lindau tumor-suppressor gene pVHL.22 Degradation of HIF alpha prevents formation of the HIF heterodimers. HIF therefore cannot bind to the hypoxia-response elements, and erythropoietin gene transcription does not occur.23
Thus, in states of hypoxia, erythropoietin production is upregulated, whereas with normal oxygen tension, production is downregulated.
Erythropoietin is essential for terminal maturation of erythrocytes
Erythropoietin is essential for terminal maturation of erythrocytes.24 It is thought to stimulate the growth of erythrogenic progenitors: burst-forming units-erythroid (BFU-E) and colony-forming units-erythroid (CFU-E). In the absence of erythropoietin, BFU-E and CFU-E fail to differentiate into mature erythrocytes.25
Binding of erythropoietin to its receptor sets off a series of downstream signals, the most important being the signal transducer and activator of transcription 5 (STAT5). In animal studies, STAT5 was found to inhibit apoptosis through the early induction of an antiapoptotic gene, Bcl-xL.26
Iron metabolism is controlled by several proteins
Iron is characterized by its capacity to accept or donate electrons. This unique property makes it a crucial element in many biochemical reactions such as enzymatic activity, DNA synthesis, oxygen transport, and cell respiration.
Iron metabolism is under the control of several proteins that play different roles in its absorption, recycling, and loss (Figure 2).27
Dietary iron exists primarily in its poorly soluble trivalent ferric form (Fe3+), and it needs to be reduced to its soluble divalent ferrous form (Fe2+) by ferric reductase to be absorbed. Ferrous iron is taken up at the apical side of enterocytes by a divalent metal transporter (DMT1) and is transported across the brush border.28
To enter the circulation, iron has to be transported across the basolateral membrane by a transporter called ferroportin.29 Ferroportin is also found in placental syncitiotrophoblasts, where it transfers iron from mother to fetus, and in macrophages, where it allows recycling of iron scavenged from damaged cells back into the circulation.30 Upon its release, the ferrous iron is oxidized to the ferric form and loaded onto transferrin. This oxidation process involves hephaestin, a homologue of the ferroxidase ceruloplasmin.31
In the plasma, iron is bound to transferrin, and under normal circumstances one-third of transferrin is saturated with iron.32 Transferrin receptors are present on most cells but are most dense on erythroid precursors. Each transferrin receptor can bind two transferrin molecules. After binding to transferrin, the transferrin receptor is endocytosed, and the iron is released into acidified vacuoles. The transferrin-receptor complex is then recycled to the surface.33
Ferritin is the cellular storage protein for iron, and it can store up to 4,500 atoms of iron within its spherical cavity.34 The serum level of ferritin reflects overall storage, with 1 ng/mL of ferritin indicating 10 mg of total iron stores.35 Ferritin is also an acute-phase reactant, and plasma levels can increase in inflammatory states such as infection or malignancy. As such, elevated ferritin does not necessarily indicate elevated iron stores.
Iron is lost in sweat, shed skin cells, and sloughed intestinal mucosal cells. However, there is no specific mechanism of iron excretion from the human body. Thus, iron is mainly regulated at the level of intestinal absorption. The iron exporter ferroportin is upregulated by the amount of available iron and is degraded by hepcidin.36
Hepcidin is a small cysteine-rich cationic peptide that is primarily produced in the liver, with some minor production also occurring in the kidneys.37 Transcription of the gene encoding hepcidin is downregulated by anemia and hypoxia and upregulated by inflammation and elevated iron levels.38 Transcription of hepcidin leads to degradation of ferroportin and a decrease in intestinal iron absorption. On the other hand, anemia and hypoxia inhibit hepcidin transcription, which allows ferroportin to facilitate intestinal iron absorption.
TREATMENT OF RENAL ANEMIA
Early enthusiasm for erythropoietin agents
Androgens started to be used to treat anemia of end-stage renal disease in 1970,39,40 and before the advent of recombinant human erythropoietin, they were a mainstay of nontransfusional therapy for anemic patients on dialysis.
The approval of recombinant human erythropoietin in 1989 drastically shifted the treatment of renal anemia. While the initial goal of treating anemia of chronic kidney disease with erythropoietin was to prevent blood transfusions,41 subsequent studies showed that the benefits might be far greater. Indeed, an initial observational trial showed that erythropoiesis-stimulating agents (ESAs) were associated with improved quality of life,42 improved neurocognitive function,43,44 and even cost savings.45 The benefits also extended to major outcomes such as regression of left ventricular hypertrophy,46 improvement in New York Heart Association class and cardiac function,47 fewer hospitalizations,48 and even reduction of cardiovascular mortality rates.49
As a result, ESA use gained popularity, and by 2006 an estimated 90% of dialysis patients were receiving these agents.50 The target and achieved hemoglobin levels also increased, with mean hemoglobin levels in hemodialysis patients being raised from 9.7 to 12 g/dL.51
Disappointing results in clinical trials of ESAs to normalize hemoglobin
To prospectively study the effects of normalized hemoglobin targets, four randomized controlled trials were conducted (Table 1):
- The Normal Hematocrit Study (NHCT)52
- The Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial53
- The Cardiovascular Risk Reduction by Early Anemia Treatment (CREATE) trial54
- The Trial to Reduce Cardiovascular Events With Aranesp Therapy (TREAT).55
These trials randomized patients to either higher “normal-range” hemoglobin targets or to lower target hemoglobin levels.
Their findings were disappointing and raised several red flags about excessive use of ESAs. The trials found no benefit in higher hemoglobin targets, and in fact, some of them demonstrated harm in patients randomized to higher targets. Notably, higher hemoglobin targets were associated with significant side effects such as access-site thrombosis,52 strokes,55 and possibly cardiovascular events.54,55 Only the CREATE trial was able to demonstrate a quality-of-life benefit for the high-target group.54
It remains unclear whether these adverse events were from the therapy itself or from an increased morbidity burden in the treated patients. Erythropoietin use is associated with hypertension,56 thought to be related to endothelin-mediated vasoconstriction.57 In our experience, this is most evident when hemoglobin levels are normalized with ESA therapy. Cycling of erythropoietin levels between extreme levels can lead to vascular remodeling, which may also be related to its cardiovascular effects.57
A noticeable finding in several of these trials was that patients failed to achieve the higher hemoglobin target despite the use of very high doses of ESA. Reanalysis of data from the CHOIR and CREATE trials showed that the patients who had worse outcomes were more likely to have required very high doses without achieving their target hemoglobin.58,59 Indeed, patients who achieved the higher target hemoglobin levels, usually at lower ESA doses, had better outcomes. This suggested that the need for a higher dose was associated with poorer outcomes, either as a marker of comorbidity or due to yet undocumented side effects of such high doses.
General approach to therapy
Before attributing anemia to chronic kidney disease, a thorough evaluation should be conducted to look for any reversible process that could be contributing to the anemia.
The causes of anemia are numerous and beyond the scope of this review. However, among the common causes of anemia in chronic kidney disease are deficiencies of iron, vitamin B12, and folate. Therefore, guidelines recommend checking iron, vitamin B12, and folate levels in the initial evaluation of anemia.60
Iron deficiency in particular is very common in chronic kidney disease patients and is present in nearly all dialysis patients.61 Hemodialysis patients are estimated to lose 1 to 3 g of iron per year as a result of blood loss in the dialysis circuit and increased iron utilization secondary to ESA therapy.62
However, in contrast to the general population, in which the upper limits of normal for iron indices are well defined, high serum ferritin levels appear to be poorly predictive of hemoglobin responsiveness in dialysis patients.63,64 Thus, the cutoffs that define iron responsiveness are much higher than standard definitions for iron deficiency.65,66 The Dialysis Patients’ Response to IV Iron With Elevated Ferritin (DRIVE) study showed that dialysis patients benefit from intravenous iron therapy even if their ferritin is as high as 1,200 ng/mL, provided their transferrin saturation is below 30%.67
Of note, erythropoietin levels cannot be used to distinguish renal anemia from other causes of anemia. Indeed, patients with renal failure may have “relative erythropoietin deficiency,” ie, “normal” erythropoietin levels that are actually too low in view of the degree of anemia.68,69 In addition to the decreased production capacity by the kidney, there appears to be a component of resistance to the action of erythropoietin in the bone marrow.
For these reasons, there is no erythropoietin level that can be considered “inadequate” or defining of renal anemia. Thus, measuring erythropoietin levels is not routinely recommended in the evaluation of renal anemia.
Two ESA preparations
The two ESAs that have traditionally been used in the treatment of renal anemia are recombinant human erythropoietin and darbepoietin alfa. They appear to be equivalent in terms of safety and efficacy.70 However, darbepoietin alfa has more sialic acid molecules, giving it a higher potency and longer half-life and allowing for less-frequent injections.71,72
In nondialysis patients, recombinant human erythropoietin is typically given every 1 to 2 weeks, whereas darbepoietin alfa can be given every 2 to 4 weeks. In dialysis patients, recombinant human erythropoietin is typically given 3 times per week with every dialysis treatment, while darbepoietin alfa is given once a week.
Target hemoglobin levels: ≤ 11.5 g/dL
In light of the four trials described in Table 1, the international Kidney Disease: Improving Global Outcomes (KDIGO) guidelines60 recommend the following (Table 2):
For patients with chronic kidney disease who are not on dialysis, ESA therapy should not be initiated if the hemoglobin level is higher than 10 g/dL. If the hemoglobin level is lower than 10 g/dL, ESA therapy can be initiated, but the decision needs to be individualized based on the rate of fall of hemoglobin concentration, prior response to iron therapy, the risk of needing a transfusion, the risks related to ESA therapy, and the presence of symptoms attributable to anemia.
For patients on dialysis, ESA therapy should be used when the hemoglobin level is between 9 and 10 g/dL to avoid having the hemoglobin fall below 9 g/dL.
In all adult patients, ESAs should not be used to intentionally increase the hemoglobin level above 13 g/dL but rather to maintain levels no higher than 11.5 g/dL. This target is based on the observation that adverse outcomes were associated with ESA use with hemoglobin targets higher than 13 g/dL (Table 1).
Target iron levels
Regarding iron stores, the guidelines recommend the following:
For adult patients with chronic kidney disease who are not on dialysis, iron should be given to keep transferrin saturation above 20% and ferritin above 100 ng/mL. Transferrin saturation should not exceed 30%, and ferritin levels should not exceed 500 ng/mL.
For adult patients on dialysis, iron should be given to maintain transferrin saturation above 30% and ferritin above 200 ng/mL.
The upper limits of ferritin and transferrin saturation are somewhat controversial, as the safety of intentionally maintaining respective levels greater than 30% and 500 ng/mL has been studied in very few patients. Transferrin saturation should in general not exceed 50%.
High ferritin levels are associated with higher death rates, but whether elevation of ferritin levels is a marker of excessive iron administration rather than a nonspecific acute-phase reactant is not clear. The 2006 guidelines60 cited upper ferritin limits of 500 to 800 ng/mL. However, the more recent DRIVE trial67 showed that patients with ferritin levels of 500 to 1,200 ng/mL will respond to intravenous administration of iron with an increase in their hemoglobin levels. This has led many clinicians to adopt a higher ferritin limit of 1,200 ng/mL.
Hemosiderosis, or excess iron deposition, was a known consequence of frequent transfusions in patients with end-stage renal disease before ESA therapy was available. However, there have been no documented cases of clinical iron overload from iron therapy using current guidelines.73
These algorithms are nuanced, and the benefit of giving intravenous iron should always be weighed against the risks of short-term acute toxicity and infection. Treatment of renal anemia not only requires in-depth knowledge of the topic, but also familiarity with the patient’s specific situation. As such, it is not recommended that clinicians unfamiliar with the treatment of renal anemia manage its treatment.
PARTICULAR CIRCUMSTANCES
Inflammation and ESA resistance
While ESAs are effective in treating anemia in many cases, in many patients the anemia fails to respond. This is of particular importance, since ESA hyporesponsiveness has been found to be a powerful predictor of cardiovascular events and death.74 It is unclear, however, whether high doses of ESA are inherently toxic or whether hyporesponsiveness is a marker of adverse outcomes related to comorbidities.
KDIGO defines initial hyporesponsiveness as having no increase in hemoglobin concentration after the first month of appropriate weight-based dosing, and acquired hyporesponsiveness as requiring two increases in ESA doses up to 50% beyond the dose at which the patient had originally been stable.60 Identifying ESA hyporesponsiveness should lead to an intensive search for potentially correctable factors.
The two major factors accounting for the state of hyporesponsiveness are inflammation and iron deficiency.75,76
Inflammation. High C-reactive protein levels have been shown to predict resistance to erythropoietin in dialysis patients.77 The release of cytokines such as tumor necrosis factor alpha, interleukin 1, and interferon gamma has an inhibitory effect on erythropoiesis.78 Additionally, inflammation can alter the response to ESAs by disrupting the metabolism of iron79 through the release of hepcidin, as previously discussed.38 These reasons likely account for the observed lower response to ESAs in the setting of acute illness and explain why ESAs are not recommended for correcting acute anemia.80
Iron deficiency also can blunt the response to ESAs. Large amounts of iron are needed for effective erythropoietic bursts. As such, iron supplementation is now a recognized treatment of renal anemia.81
Other factors associated with hyporesponsiveness include chronic occult blood loss, aluminum toxicity, cobalamin or folate deficiencies, testosterone deficiency, inadequate dialysis, hyperparathyroidism, and superimposed primary bone marrow disease,82,83 and these should be addressed in patients whose anemia does not respond as expected to ESA therapy. A summary of the main causes of ESA hyporesponsiveness, their reversibility, and recommended treatments is presented in Table 3.
Antibody-mediated pure red-cell aplasia. Rarely, patients receiving ESA therapy develop antibodies that neutralize both the ESA and endogenous erythropoietin. The resulting syndrome, called antibody-mediated pure red-cell aplasia, is characterized by the sudden development of severe transfusion-dependent anemia. This has historically been connected to epoetin beta, a formulation not in use in the United States. However, cases have been documented with epoetin alfa and darbepoetin. The incidence rate is low with subcutaneous ESA use, estimated at 0.5 cases per 10,000 patient-years84 and anecdotal with intravenous ESA.85 The definitive diagnosis requires demonstration of neutralizing antibodies against erythropoietin. Parvovirus infection should be excluded as an alternative cause of pure redcell aplasia.
ANEMIA IN CANCER PATIENTS
ESAs are effective in raising hemoglobin levels and reducing transfusion requirements in patients with chemotherapy-induced anemia.86 However, there are data linking the use of ESAs to shortened survival in patients who have a variety of solid tumors.87
Several mechanisms have been proposed to explain this rapid disease progression, most notably acceleration in tumor growth88–90 by stimulation of erythropoietin receptors on the surface of the tumor cells, leading to increased tumor angiogenesis.91,92
For these reasons, treatment of renal anemia in the setting of active malignancy should be referred to an oncologist.
NOVEL TREATMENTS
Several new agents for treating renal anemia are currently under review.
Continuous erythropoiesis receptor activator
Continuous erythropoiesis receptor activator is a pegylated form of recombinant human erythropoietin that has the ability to repeatedly activate the erythropoietin receptor. It appears to be similar to the other forms of erythropoietin in terms of safety and efficacy in both end-stage renal disease93 and chronic kidney disease.94 It has the advantage of an extended serum half-life, which allows for longer dosing intervals, ie, every 2 weeks. Its use is currently gaining popularity in the dialysis community.
HIF stabilizers
Our growing understanding of the physiology of erythropoietin offers new potential treatment targets. As previously described, production of erythropoietin is stimulated by HIFs. In order to be degraded, these HIFs are hydroxylated at their proline residues by a prolyl hydroxylase. A new category of drugs called prolyl-hydroxylase inhibitors (PDIs) offers the advantage of stabilizing the HIFs, leading to an increase in erythropoietin production.
In phase 1 and 2 clinical trials, these agents have been shown to increase hemoglobin in both end-stage renal disease and chronic kidney disease patients15,16 but not in anephric patients, demonstrating a renal source of the erythropoietin production even in nonfunctioning kidneys. The study of one PDI agent (FG 2216) was halted temporarily after a report of death from fulminant hepatitis, but the other (FG 4592) continues to be studied in a phase 2 clinical trial.95,96
TAKE-HOME POINTS
- Anemia of renal disease is a common condition that is mainly caused by a decrease in erythropoietin production by the kidneys.
- While anemia of renal disease can be corrected with ESAs, it is necessary to investigate and rule out underlying treatable conditions such as iron or vitamin deficiencies before giving an ESA.
- Anemia of renal disease is associated with significant morbidity such as increased risk of left ventricular hypertrophy, myocardial infarction, and heart failure, and has been described as an all-cause mortality multiplier.
- Unfortunately, the only undisputed benefit of treatment to date remains the avoidance of blood transfusions. Furthermore, the large randomized controlled trials that looked at the benefits of ESA have shown that their use can be associated with increased risk of cardiovascular events. Therefore, use of an ESA in end-stage renal disease should never target a normal hemoglobin levels but rather aim for a hemoglobin level of no more than 11.5 g/dL.
- Use of an ESA in chronic kidney disease should be individualized and is not recommended to be started unless the hemoglobin level is less than 10 g/dL.
- Several newer agents for renal anemia are currently under review. A pegylated form of recombinant human erythropoietin has an extended half-life, and a new and promising category of drugs called HIF stabilizers is currently under study.
- World Health Organization (WHO). Nutritional anaemias: report of a WHO scientific group. Geneva, Switzerland: World Health Organization, 1968.
- Hsu CY, McCulloch CE, Curhan GC, et al. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol 2002; 13:504–510.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoetin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Kazmi WH, Kausz AT, Khan S, et al. Anemia: an early complication of chronic renal insufficiency. Am J Kidney Dis 2001; 38:803–812.
- United States Renal Data System. Chapter 3. Morbidity & mortality in patients with CKD. www.usrds.org/2012/view/v1_03.aspx. Accessed June 9, 2016.
- Horwich TB, Fonarow GC, Hamilton MA, MacLellan WR, Borenstein J. Anemia is associated with worse symptoms, greater impairment in functional capacity and a significant increase in mortality in patients with advanced heart failure. J Am Coll Cardiol 2002; 39:1780–1786.
- Mark DB, Felker GM. B-type natriuretic peptide: a biomarker for all seasons? N Engl J Med 2004; 350:718–720.
- Walker AM, Schneider G, Yeaw J, Nordstrom B, Robbins S, Pettitt D. Anemia as a predictor of cardiovascular events in patients with elevated serum creatinine. J Am Soc Nephrol 2006; 17:2293–2298.
- Abramson JL, Jurkovitz CT, Vaccarino V, Weintraub WS, McClellan W. Chronic kidney disease, anemia, and incident stroke in a middle-aged, community-based population: the ARIC Study. Kidney Int 2003; 64:610–615.
- Sarnak MJ, Tighiouart H, Manjunath G, et al. Anemia as a risk factor for cardiovascular disease in the Atherosclerosis Risk in Communities (ARIC) study. J Am Coll Cardiol 2002; 40:27–33.
- McClellan WM, Flanders WD, Langston RD, Jurkovitz C, Presley R. Anemia and renal insufficiency are independent risk factors for death among patients with congestive heart failure admitted to community hospitals: a population-based study. J Am Soc Nephrol 2002; 13:1928–1936.
- Xia H, Ebben J, Ma JZ, Collins AJ. Hematocrit levels and hospitalization risks in hemodialysis patients. J Am Soc Nephrol 1999; 10:1309–1316.
- Collins AJ, Li S, St Peter W, et al. Death, hospitalization, and economic associations among incident hemodialysis patients with hematocrit values of 36 to 39%. J Am Soc Nephrol 2001; 12:2465–2473.
- Agarwal AK. Practical approach to the diagnosis and treatment of anemia associated with CKD in elderly. J Am Med Dir Assoc 2006; 7(suppl 9):S7–S12.
- Bernhardt WM, Wiesener MS, Scigalla P, et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol 2010; 21:2151–2156.
- Provenzano R, Fadda G, Bernardo M, et al. FG-2216, a novel oral HIF-PHI, stimulates erythropoiesis and increases hemoglobin concentration in patients with non-dialysis CKD. Am J Kidney Dis 2008; 51:B80.
- Maxwell PH, Osmond MK, Pugh CW, et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int 1993; 44:1149–1162.
- Maxwell PH, Ferguson DJ, Nicholls LG, et al. Sites of erythropoietin production. Kidney Int 1997; 51:393–401.
- Jelkmann W. Erythropoeitin: structure, control of production and function. Physiol Rev 1992; 72:449–489.
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92:5510–5514.
- Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 1995; 270:1230–1237.
- Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399:271–275.
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 1997; 272:22642–22647.
- Malik J, Kim AR, Tyre KA, Cherukuri AR, Palis J. Erythropoietin critically regulates the terminal maturation of murine and human primitive erythroblasts. Haematologica 2013; 98:1778–1787.
- Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 1995; 83:59–67.
- Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(-/-)5b(-/-) mice due to decreased survival of early erythroblasts. Blood 2001; 98:3261–3273.
- Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol 2005; 202:199–211.
- Conrad ME, Umbreit JN. Pathways of iron absorption. Blood Cells Mol Dis 2002; 29:336–355.
- Frazer DM, Anderson GJ. The orchestration of body iron intake: how and where do enterocytes receive their cues? Blood Cells Moll Dis 2003; 30:288–297.
- Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 2005; 1:191–200.
- Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999; 21:195–199.
- Bothwell TH. Overview and mechanisms of iron regulation. Nutr Rev 1995: 53:237–245.
- Kawabata H, Nakamaki T, Ikonomi P, Smith RD, Germain RS, Koeffler HP. Expression of transferrin receptor 2 in normal and neoplastic hematopoietic cells. Blood 2001; 98:2714–2719.
- Arosio P, Levi S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim Biophys Acta 2010; 1800:783–792.
- Finch CA, Bellotti V, Stray S, et al. Plasma ferritin determination as a diagnostic tool. West J Med 1986; 145:657–663.
- Delaby C, Pilard N, Goncalves AS, Beaumont C, Canonne-Hergaux F. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 2005; 106:3979–3984.
- Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003; 102:783–788.
- Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002; 110:1037–1044.
- DeGowin RL, Lavender AR, Forland M, Charleston D, Gottschalk A. Erythropoiesis and erythropoietin in patients with chronic renal failure treated with hemodialysis and testosterone. Ann Intern Med 1970; 72:913–918.
- Richardson JR Jr, Weinstein MB. Erythropoietic response of dialyzed patients to testosterone administration. Ann Intern Med 1970; 73:403–407
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Intern Med 1989; 111:992–1000.
- Moreno F, Aracil FJ, Pérez R, Valderrábano F. Controlled study on the improvement of quality of life in elderly hemodialysis patients after correcting end-stage renal disease-related anemia with erythropoietin. Am J Kidney Dis 1996; 27:548–556.
- Nissenson AR, Nimer SD, Wolcott DL. Recombinant human erythropoietin and renal anemia: molecular biology, clinical efficacy, and nervous system effects. Ann Intern Med 1991; 114:402–416.
- Stivelman JC. Benefits of anaemia treatment on cognitive function. Nephrol Dial Transplant 2000; 15(suppl 3):29–35.
- Maddux FW, Shetty S, del Aguila MA, Nelson MA, Murray BM. Effect of erythropoiesis-stimulating agents on healthcare utilization, costs, and outcomes in chronic kidney disease. Ann Pharmacother 2007; 41:1761–1769.
- Macdougall IC, Lewis NP, Saunders MJ, et al. Long-term cardiorespiratory effects of amelioration of renal anaemia by erythropoietin. Lancet 1990; 335:489–493.
- Silverberg DS, Wexler D, Blum M, et al. Effects of treatment with epoetin beta on outcomes in patients with anaemia and chronic heart failure. Kidney Blood Press Res 2005; 28:41–47.
- Perkins R, Olson S, Hansen J, Lee J, Stiles K, Lebrun C. Impact of an anemia clinic on emergency room visits and hospitalizations in patients with anemia of CKD pre-dialysis. Nephrol Nurs J 2007; 34:167–173, 182.
- Locatelli F, Conte F, Marcelli D. The impact of haematocrit levels and erythropoietin treatment on overall and cardiovascular mortality and morbidity—the experience of the Lombardy Dialysis Registry. Nephrol Dial Transplant 1998; 13:1642–1644.
- Centers for Medicare and Medicaid Services; Kinney R. 2005 Annual Report: ESRD Clinical Performance Measures Project. Am J Kidney Dis 2006; 48(suppl 2):S1–S106.
- US Renal Data System. Annual Data Report 2006. www.usrds.org/adr.aspx. Accessed July 3, 2016.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590.
- Singh AK, Szczech L, Tang KL, et al; CHOIR Investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- Pfeffer MA, Burdmann EA, Chen CY, et al; TREAT Investigators. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 2009; 361:2019–2032.
- Kirkpantur A, Kahraman S, Yilmaz R, et al. The effects of maintenance recombinant human erythropoietin therapy on ambulatory blood pressure recordings: conventional, Doppler, and tissue Doppler echocardiographic parameters. Artif Organs 2005; 29:965–972.
- Fishbane S, Berns JS. Hemoglobin cycling in hemodialysis patients treated with recombinant human erythropoietin. Kidney Int 2005; 68:1337–1343.
- Szczech LA, Barnhart HX, Inrig JK, et al. Secondary analysis of the CHOIR trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int 2008; 74:791–798.
- Solomon SD, Uno H, Lewis EF, et al; Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) Investigators. Erythropoietic response and outcomes in kidney disease and type 2 diabetes. N Engl J Med 2010; 363:1146–1155.
- Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney Int Suppl 2012; 2:279–335.
- Fernández-Rodríguez AM, Guindeo-Casasús MC, Molero-Labarta T, et al. Diagnosis of iron deficiency in chronic renal failure. Am J Kidney Dis 1999; 34:508–513.
- Eschbach JW, Cook JD, Scribner BH, Finch CA. Iron balance in hemodialysis patients. Ann Intern Med 1977; 87:710–713.
- Mittman N, Sreedhara R, Mushnick R, et al. Reticulocyte hemoglobin content predicts functional iron deficiency in hemodialysis patients receiving rHuEPO. Am J Kidney Dis 1997; 30:912–922.
- Tessitore N, Solero GP, Lippi G, et al. The role of iron status markers in predicting response to intravenous iron in haemodialysis patients on maintenance erythropoietin. Nephrol Dial Transplant 2001; 16:1416–1423.
- Coyne DW. Iron indices: what do they really mean? Kidney Int Suppl 2006; 101:S4–S8.
- Fishbane S, Kowalski EA, Imbriano LJ, Maesaka JK. The evaluation of iron status in hemodialysis patients. J Am Soc Nephrol 1996; 7:2654–2657.
- Coyne DW, Kapoian T, Suki W, et al; DRIVE Study Group. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: results of the Dialysis Patients’ Response to IV Iron with Elevated Ferritin (DRIVE) Study. J Am Soc Nephrol 2007; 18:975–984.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoietin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Korte W, Cogliatti SB, Jung K, Riesen W. Mild renal dysfunction is sufficient to induce erythropoietin deficiency in patients with unexplained anaemia. Clin Chim Acta 2000; 292:149–154.
- Locatelli F, Olivares J, Walker R, et al; European/Australian NESP 980202 Study Group. Novel erythropoiesis stimulating protein for treatment of anemia in chronic renal insufficiency. Kidney Int 2001; 60:741–747.
- Carrera F, Burnier M. Use of darbepoetin alfa in the treatment of anaemia of chronic kidney disease: clinical and pharmacoeconomic considerations. NDT Plus 2009; 2(suppl 1):i9–i17.
- Egrie JC, Browne JK. Development and characterization of novel erythropoiesis stimulating protein (NESP). Nephrol Dial Transplant 2001; 16(suppl 3):3–13.
- Nissenson AR, Charytan C. Controversies in iron management. Kidney Int Suppl 2003; 87:S64–S71.
- Kilpatrick RD, Critchlow CW, Fishbane S, et al. Greater epoetin alpha responsiveness is associated with improved survival in hemodialysis patients. Clin J Am Soc Nephrol 2008; 3:1077–1083.
- Locatelli F, Aljama P, Barany P, et al; European Best Practice Guidelines Working Group. Revised European best practice guidelines for the management of anaemia in patients with chronic renal failure. Nephrol Dial Transplant 2004; 19(suppl 2):ii1–ii47.
- Stenvinkel P. The role of inflammation in the anaemia of end-stage renal disease. Nephrol Dial Transplant 2001; 16(suppl 7):36–40.
- Barany P, Divino Filho JC, Bergstrom J. High C-reactive protein is a strong predictor of resistance to erythropoietin in hemodialysis patients. Am J Kidney Dis 1997; 29:565–568.
- Drueke T. Hyporesponsiveness to recombinant human erythropoietin. Nephrol Dial Transplant 2001; 16(suppl 7):25–28.
- Casadevall N. Cellular mechanism of resistance to erythropoietin. Nephrol Dial Transplant 1995; 10(suppl 6):27–30.
- Kraus E, Rabb H. EPO therapy during acute kidney disease: to use or not to use, that is the question. Am J Kidney Dis 2005; 46:967–969.
- Gotloib L, Silverberg D, Fudin R, Shostak A. Iron deficiency is a common cause of anemia in chronic kidney disease and can often be corrected with intravenous iron. J Nephrol 2006; 19:161–167.
- Tarng DC, Huang TP, Chen TW, Yang WC. Erythropoietin hyporesponsiveness: from iron deficiency to iron overload. Kidney Int Suppl 1999; 69:S107–S118.
- Drüeke TB. Modulating factors in the hematopoietic response to erythropoietin. Am J Kidney Dis 1991; 18(suppl 1):87–92.
- Boven K, Stryker S, Knight J, et al. The increased incidence of pure red cell aplasia with an Eprex formulation in uncoated rubber stopper syringes. Kidney Int 2005; 67:2346–2353.
- Shimizu H, Saitoh T, Ota F, et al. Pure red cell aplasia induced only by intravenous administration of recombinant human erythropoietin. Acta Haematol 2011; 126:114–118.
- Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst Rev 2012; 12:CD003407.
- Bohlius J, Langensiepen S, Schwarzer G, et al. Recombinant human erythropoietin and overall survival in cancer patients: results of a comprehensive meta-analysis. J Natl Cancer Inst 2005; 97:489–498.
- Henke M, Laszig R, Rübe C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet 2003; 362:1255–1260.
- Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005; 23:5960–5972.
- Brower V. Erythropoietin may impair, not improve, cancer survival. Nat Med 2003; 9:1439.
- Acs G, Acs P, Beckwith SM, et al. Erythropoietin and erythropoietin receptor expression in human cancer. Cancer Res 2001; 61:3561–3565.
- Yasuda Y, Fujita Y, Matsuo T, et al. Erythropoietin regulates tumour growth of human malignancies. Carcinogenesis 2003; 24:1021–1029.
- Levin NW, Fishbane S, Cañedo FV, et al; MAXIMA Study Investigators. Intravenous methoxy polyethylene glycol-epoetin beta for haemoglobin control in patients with chronic kidney disease who are on dialysis: a randomised non-inferiority trial (MAXIMA). Lancet 2007; 370:1415–1421.
- Macdougall IC, Walker R, Provenzano R, et al; ARCTOS Study Investigators. C.E.R.A. corrects anemia in patients with chronic kidney disease not on dialysis: results of a randomized clinical trial. Clin J Am Soc Nephrol 2008; 3:337–347.
- Frohna PA, Milwee S, Pinkett J, et al. Preliminary results from a randomized, single-blind, placebo-controlled trial of FG-4592, a novel hypoxia inducible factor prolyl hydroxylase inhibitor, in subjects with CKD anemia (abstract). J Am Soc Nephrol 2007; 18:763.
- Holdstock L, Meadowcroft AM, Maier R, et al. Four-week studies of oral hypoxia-inducible factor-prolyl hydroxylase inhibitor GSK1278863 for treatment of anemia. J Am Soc Nephrol 2016; 27:1234–1244.
- World Health Organization (WHO). Nutritional anaemias: report of a WHO scientific group. Geneva, Switzerland: World Health Organization, 1968.
- Hsu CY, McCulloch CE, Curhan GC, et al. Epidemiology of anemia associated with chronic renal insufficiency among adults in the United States: results from the Third National Health and Nutrition Examination Survey. J Am Soc Nephrol 2002; 13:504–510.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoetin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Kazmi WH, Kausz AT, Khan S, et al. Anemia: an early complication of chronic renal insufficiency. Am J Kidney Dis 2001; 38:803–812.
- United States Renal Data System. Chapter 3. Morbidity & mortality in patients with CKD. www.usrds.org/2012/view/v1_03.aspx. Accessed June 9, 2016.
- Horwich TB, Fonarow GC, Hamilton MA, MacLellan WR, Borenstein J. Anemia is associated with worse symptoms, greater impairment in functional capacity and a significant increase in mortality in patients with advanced heart failure. J Am Coll Cardiol 2002; 39:1780–1786.
- Mark DB, Felker GM. B-type natriuretic peptide: a biomarker for all seasons? N Engl J Med 2004; 350:718–720.
- Walker AM, Schneider G, Yeaw J, Nordstrom B, Robbins S, Pettitt D. Anemia as a predictor of cardiovascular events in patients with elevated serum creatinine. J Am Soc Nephrol 2006; 17:2293–2298.
- Abramson JL, Jurkovitz CT, Vaccarino V, Weintraub WS, McClellan W. Chronic kidney disease, anemia, and incident stroke in a middle-aged, community-based population: the ARIC Study. Kidney Int 2003; 64:610–615.
- Sarnak MJ, Tighiouart H, Manjunath G, et al. Anemia as a risk factor for cardiovascular disease in the Atherosclerosis Risk in Communities (ARIC) study. J Am Coll Cardiol 2002; 40:27–33.
- McClellan WM, Flanders WD, Langston RD, Jurkovitz C, Presley R. Anemia and renal insufficiency are independent risk factors for death among patients with congestive heart failure admitted to community hospitals: a population-based study. J Am Soc Nephrol 2002; 13:1928–1936.
- Xia H, Ebben J, Ma JZ, Collins AJ. Hematocrit levels and hospitalization risks in hemodialysis patients. J Am Soc Nephrol 1999; 10:1309–1316.
- Collins AJ, Li S, St Peter W, et al. Death, hospitalization, and economic associations among incident hemodialysis patients with hematocrit values of 36 to 39%. J Am Soc Nephrol 2001; 12:2465–2473.
- Agarwal AK. Practical approach to the diagnosis and treatment of anemia associated with CKD in elderly. J Am Med Dir Assoc 2006; 7(suppl 9):S7–S12.
- Bernhardt WM, Wiesener MS, Scigalla P, et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol 2010; 21:2151–2156.
- Provenzano R, Fadda G, Bernardo M, et al. FG-2216, a novel oral HIF-PHI, stimulates erythropoiesis and increases hemoglobin concentration in patients with non-dialysis CKD. Am J Kidney Dis 2008; 51:B80.
- Maxwell PH, Osmond MK, Pugh CW, et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int 1993; 44:1149–1162.
- Maxwell PH, Ferguson DJ, Nicholls LG, et al. Sites of erythropoietin production. Kidney Int 1997; 51:393–401.
- Jelkmann W. Erythropoeitin: structure, control of production and function. Physiol Rev 1992; 72:449–489.
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 1995; 92:5510–5514.
- Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem 1995; 270:1230–1237.
- Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399:271–275.
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 1997; 272:22642–22647.
- Malik J, Kim AR, Tyre KA, Cherukuri AR, Palis J. Erythropoietin critically regulates the terminal maturation of murine and human primitive erythroblasts. Haematologica 2013; 98:1778–1787.
- Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell 1995; 83:59–67.
- Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(-/-)5b(-/-) mice due to decreased survival of early erythroblasts. Blood 2001; 98:3261–3273.
- Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol 2005; 202:199–211.
- Conrad ME, Umbreit JN. Pathways of iron absorption. Blood Cells Mol Dis 2002; 29:336–355.
- Frazer DM, Anderson GJ. The orchestration of body iron intake: how and where do enterocytes receive their cues? Blood Cells Moll Dis 2003; 30:288–297.
- Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab 2005; 1:191–200.
- Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999; 21:195–199.
- Bothwell TH. Overview and mechanisms of iron regulation. Nutr Rev 1995: 53:237–245.
- Kawabata H, Nakamaki T, Ikonomi P, Smith RD, Germain RS, Koeffler HP. Expression of transferrin receptor 2 in normal and neoplastic hematopoietic cells. Blood 2001; 98:2714–2719.
- Arosio P, Levi S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim Biophys Acta 2010; 1800:783–792.
- Finch CA, Bellotti V, Stray S, et al. Plasma ferritin determination as a diagnostic tool. West J Med 1986; 145:657–663.
- Delaby C, Pilard N, Goncalves AS, Beaumont C, Canonne-Hergaux F. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 2005; 106:3979–3984.
- Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003; 102:783–788.
- Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002; 110:1037–1044.
- DeGowin RL, Lavender AR, Forland M, Charleston D, Gottschalk A. Erythropoiesis and erythropoietin in patients with chronic renal failure treated with hemodialysis and testosterone. Ann Intern Med 1970; 72:913–918.
- Richardson JR Jr, Weinstein MB. Erythropoietic response of dialyzed patients to testosterone administration. Ann Intern Med 1970; 73:403–407
- Eschbach JW, Abdulhadi MH, Browne JK, et al. Recombinant human erythropoietin in anemic patients with end-stage renal disease. Results of a phase III multicenter clinical trial. Ann Intern Med 1989; 111:992–1000.
- Moreno F, Aracil FJ, Pérez R, Valderrábano F. Controlled study on the improvement of quality of life in elderly hemodialysis patients after correcting end-stage renal disease-related anemia with erythropoietin. Am J Kidney Dis 1996; 27:548–556.
- Nissenson AR, Nimer SD, Wolcott DL. Recombinant human erythropoietin and renal anemia: molecular biology, clinical efficacy, and nervous system effects. Ann Intern Med 1991; 114:402–416.
- Stivelman JC. Benefits of anaemia treatment on cognitive function. Nephrol Dial Transplant 2000; 15(suppl 3):29–35.
- Maddux FW, Shetty S, del Aguila MA, Nelson MA, Murray BM. Effect of erythropoiesis-stimulating agents on healthcare utilization, costs, and outcomes in chronic kidney disease. Ann Pharmacother 2007; 41:1761–1769.
- Macdougall IC, Lewis NP, Saunders MJ, et al. Long-term cardiorespiratory effects of amelioration of renal anaemia by erythropoietin. Lancet 1990; 335:489–493.
- Silverberg DS, Wexler D, Blum M, et al. Effects of treatment with epoetin beta on outcomes in patients with anaemia and chronic heart failure. Kidney Blood Press Res 2005; 28:41–47.
- Perkins R, Olson S, Hansen J, Lee J, Stiles K, Lebrun C. Impact of an anemia clinic on emergency room visits and hospitalizations in patients with anemia of CKD pre-dialysis. Nephrol Nurs J 2007; 34:167–173, 182.
- Locatelli F, Conte F, Marcelli D. The impact of haematocrit levels and erythropoietin treatment on overall and cardiovascular mortality and morbidity—the experience of the Lombardy Dialysis Registry. Nephrol Dial Transplant 1998; 13:1642–1644.
- Centers for Medicare and Medicaid Services; Kinney R. 2005 Annual Report: ESRD Clinical Performance Measures Project. Am J Kidney Dis 2006; 48(suppl 2):S1–S106.
- US Renal Data System. Annual Data Report 2006. www.usrds.org/adr.aspx. Accessed July 3, 2016.
- Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med 1998; 339:584–590.
- Singh AK, Szczech L, Tang KL, et al; CHOIR Investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- Pfeffer MA, Burdmann EA, Chen CY, et al; TREAT Investigators. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 2009; 361:2019–2032.
- Kirkpantur A, Kahraman S, Yilmaz R, et al. The effects of maintenance recombinant human erythropoietin therapy on ambulatory blood pressure recordings: conventional, Doppler, and tissue Doppler echocardiographic parameters. Artif Organs 2005; 29:965–972.
- Fishbane S, Berns JS. Hemoglobin cycling in hemodialysis patients treated with recombinant human erythropoietin. Kidney Int 2005; 68:1337–1343.
- Szczech LA, Barnhart HX, Inrig JK, et al. Secondary analysis of the CHOIR trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int 2008; 74:791–798.
- Solomon SD, Uno H, Lewis EF, et al; Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) Investigators. Erythropoietic response and outcomes in kidney disease and type 2 diabetes. N Engl J Med 2010; 363:1146–1155.
- Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney Int Suppl 2012; 2:279–335.
- Fernández-Rodríguez AM, Guindeo-Casasús MC, Molero-Labarta T, et al. Diagnosis of iron deficiency in chronic renal failure. Am J Kidney Dis 1999; 34:508–513.
- Eschbach JW, Cook JD, Scribner BH, Finch CA. Iron balance in hemodialysis patients. Ann Intern Med 1977; 87:710–713.
- Mittman N, Sreedhara R, Mushnick R, et al. Reticulocyte hemoglobin content predicts functional iron deficiency in hemodialysis patients receiving rHuEPO. Am J Kidney Dis 1997; 30:912–922.
- Tessitore N, Solero GP, Lippi G, et al. The role of iron status markers in predicting response to intravenous iron in haemodialysis patients on maintenance erythropoietin. Nephrol Dial Transplant 2001; 16:1416–1423.
- Coyne DW. Iron indices: what do they really mean? Kidney Int Suppl 2006; 101:S4–S8.
- Fishbane S, Kowalski EA, Imbriano LJ, Maesaka JK. The evaluation of iron status in hemodialysis patients. J Am Soc Nephrol 1996; 7:2654–2657.
- Coyne DW, Kapoian T, Suki W, et al; DRIVE Study Group. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: results of the Dialysis Patients’ Response to IV Iron with Elevated Ferritin (DRIVE) Study. J Am Soc Nephrol 2007; 18:975–984.
- Radtke HW, Claussner A, Erbes PM, Scheuermann EH, Schoeppe W, Koch KM. Serum erythropoietin concentration in chronic renal failure: relationship to degree of anemia and excretory renal function. Blood 1979; 54:877–884.
- Korte W, Cogliatti SB, Jung K, Riesen W. Mild renal dysfunction is sufficient to induce erythropoietin deficiency in patients with unexplained anaemia. Clin Chim Acta 2000; 292:149–154.
- Locatelli F, Olivares J, Walker R, et al; European/Australian NESP 980202 Study Group. Novel erythropoiesis stimulating protein for treatment of anemia in chronic renal insufficiency. Kidney Int 2001; 60:741–747.
- Carrera F, Burnier M. Use of darbepoetin alfa in the treatment of anaemia of chronic kidney disease: clinical and pharmacoeconomic considerations. NDT Plus 2009; 2(suppl 1):i9–i17.
- Egrie JC, Browne JK. Development and characterization of novel erythropoiesis stimulating protein (NESP). Nephrol Dial Transplant 2001; 16(suppl 3):3–13.
- Nissenson AR, Charytan C. Controversies in iron management. Kidney Int Suppl 2003; 87:S64–S71.
- Kilpatrick RD, Critchlow CW, Fishbane S, et al. Greater epoetin alpha responsiveness is associated with improved survival in hemodialysis patients. Clin J Am Soc Nephrol 2008; 3:1077–1083.
- Locatelli F, Aljama P, Barany P, et al; European Best Practice Guidelines Working Group. Revised European best practice guidelines for the management of anaemia in patients with chronic renal failure. Nephrol Dial Transplant 2004; 19(suppl 2):ii1–ii47.
- Stenvinkel P. The role of inflammation in the anaemia of end-stage renal disease. Nephrol Dial Transplant 2001; 16(suppl 7):36–40.
- Barany P, Divino Filho JC, Bergstrom J. High C-reactive protein is a strong predictor of resistance to erythropoietin in hemodialysis patients. Am J Kidney Dis 1997; 29:565–568.
- Drueke T. Hyporesponsiveness to recombinant human erythropoietin. Nephrol Dial Transplant 2001; 16(suppl 7):25–28.
- Casadevall N. Cellular mechanism of resistance to erythropoietin. Nephrol Dial Transplant 1995; 10(suppl 6):27–30.
- Kraus E, Rabb H. EPO therapy during acute kidney disease: to use or not to use, that is the question. Am J Kidney Dis 2005; 46:967–969.
- Gotloib L, Silverberg D, Fudin R, Shostak A. Iron deficiency is a common cause of anemia in chronic kidney disease and can often be corrected with intravenous iron. J Nephrol 2006; 19:161–167.
- Tarng DC, Huang TP, Chen TW, Yang WC. Erythropoietin hyporesponsiveness: from iron deficiency to iron overload. Kidney Int Suppl 1999; 69:S107–S118.
- Drüeke TB. Modulating factors in the hematopoietic response to erythropoietin. Am J Kidney Dis 1991; 18(suppl 1):87–92.
- Boven K, Stryker S, Knight J, et al. The increased incidence of pure red cell aplasia with an Eprex formulation in uncoated rubber stopper syringes. Kidney Int 2005; 67:2346–2353.
- Shimizu H, Saitoh T, Ota F, et al. Pure red cell aplasia induced only by intravenous administration of recombinant human erythropoietin. Acta Haematol 2011; 126:114–118.
- Tonia T, Mettler A, Robert N, et al. Erythropoietin or darbepoetin for patients with cancer. Cochrane Database Syst Rev 2012; 12:CD003407.
- Bohlius J, Langensiepen S, Schwarzer G, et al. Recombinant human erythropoietin and overall survival in cancer patients: results of a comprehensive meta-analysis. J Natl Cancer Inst 2005; 97:489–498.
- Henke M, Laszig R, Rübe C, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet 2003; 362:1255–1260.
- Leyland-Jones B, Semiglazov V, Pawlicki M, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol 2005; 23:5960–5972.
- Brower V. Erythropoietin may impair, not improve, cancer survival. Nat Med 2003; 9:1439.
- Acs G, Acs P, Beckwith SM, et al. Erythropoietin and erythropoietin receptor expression in human cancer. Cancer Res 2001; 61:3561–3565.
- Yasuda Y, Fujita Y, Matsuo T, et al. Erythropoietin regulates tumour growth of human malignancies. Carcinogenesis 2003; 24:1021–1029.
- Levin NW, Fishbane S, Cañedo FV, et al; MAXIMA Study Investigators. Intravenous methoxy polyethylene glycol-epoetin beta for haemoglobin control in patients with chronic kidney disease who are on dialysis: a randomised non-inferiority trial (MAXIMA). Lancet 2007; 370:1415–1421.
- Macdougall IC, Walker R, Provenzano R, et al; ARCTOS Study Investigators. C.E.R.A. corrects anemia in patients with chronic kidney disease not on dialysis: results of a randomized clinical trial. Clin J Am Soc Nephrol 2008; 3:337–347.
- Frohna PA, Milwee S, Pinkett J, et al. Preliminary results from a randomized, single-blind, placebo-controlled trial of FG-4592, a novel hypoxia inducible factor prolyl hydroxylase inhibitor, in subjects with CKD anemia (abstract). J Am Soc Nephrol 2007; 18:763.
- Holdstock L, Meadowcroft AM, Maier R, et al. Four-week studies of oral hypoxia-inducible factor-prolyl hydroxylase inhibitor GSK1278863 for treatment of anemia. J Am Soc Nephrol 2016; 27:1234–1244.
KEY POINTS
- Before treating with ESAs, it is necessary to investigate and rule out underlying treatable conditions such as iron or vitamin deficiencies.
- Recognizing anemia in chronic kidney disease is important and often involves participation by the primary care physician, especially in early disease when chronic kidney disease may be mild.
- The only proven benefit of ESA therapy is avoidance of blood transfusions.
- ESAs should not be used to increase the hemoglobin concentration above 13 g/dL. In end-stage renal disease, the goal of therapy is to maintain levels at a target no higher than 11.5 g/dL. In nondialysis-dependent chronic kidney disease, the decision to prescribe ESA therapy should be individualized.
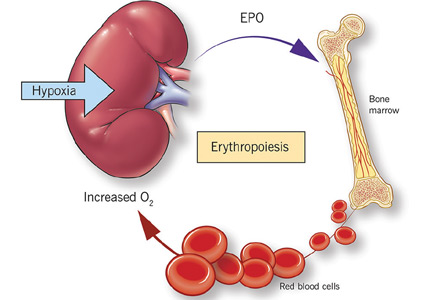
In reply: Why is metformin contraindicated in chronic kidney disease?
In Reply: We appreciate Dr. Imam’s comments regarding using metformin in those with chronic kidney disease.
The US Food and Drug Administration currently lists metformin as contraindicated in those with mild to moderate renal insufficiency, with serum creatinine levels greater than or equal to 1.5 mg/dL in males and greater than or equal to 1.4 mg/dL in females. This contraindication is based on the pharmacokinetics of the medication and, likely, the association of a similar medication, phenformin, with lactic acidosis, which eventually led to its withdrawal from the market. However, lactic acidosis is much less frequent with metformin than with phenformin.1
We agree that metformin is an invaluable medication for diabetes mellitus not requiring insulin. We also agree that lactic acidosis is rare, especially in those with mild renal insufficiency. However, lactic acidosis does occur in patients with chronic kidney disease while on metformin and, however rare, when it does occur it is a life-threatening event.2
The clearance of metformin is strongly dependent on kidney function,3 and therefore guidelines still recommend reducing the dose in those with moderate renal insufficiency and recommend considering stopping the medication in those with severe renal insufficiency—the population we were talking about in our article.4 We are aware of changes to the guidelines that have been made by various groups, and in many circumstances we ourselves take an individualized approach, weighing the risks and benefits of continued therapy with the patient and his or her primary care provider. That being said, we did not believe that such nuanced recommendations were appropriate for our article, especially since they are contrary to marketing restrictions for the drug.
- Bailey CJ, Turner RC. Metformin. N Engl J Med 1996; 334:574–579.
- Lalau JD, Race JM. Lactic acidosis in metformin-treated patients. Prognostic value of arterial lactate levels and plasma metformin concentrations. Drug Saf 1999; 20:377–384.
- Sambol NC, Chiang J, Lin ET, et al. Kidney function and age are both predictors of pharmacokinetics of metformin. J Clin Pharmacol 1995; 35:1094–1102.
- Sakhuja A, Hyland J, Simon JF. Managing advanced chronic kidney disease: a primary care guide. Cleve Clin J Med 2014; 81:289–299.
In Reply: We appreciate Dr. Imam’s comments regarding using metformin in those with chronic kidney disease.
The US Food and Drug Administration currently lists metformin as contraindicated in those with mild to moderate renal insufficiency, with serum creatinine levels greater than or equal to 1.5 mg/dL in males and greater than or equal to 1.4 mg/dL in females. This contraindication is based on the pharmacokinetics of the medication and, likely, the association of a similar medication, phenformin, with lactic acidosis, which eventually led to its withdrawal from the market. However, lactic acidosis is much less frequent with metformin than with phenformin.1
We agree that metformin is an invaluable medication for diabetes mellitus not requiring insulin. We also agree that lactic acidosis is rare, especially in those with mild renal insufficiency. However, lactic acidosis does occur in patients with chronic kidney disease while on metformin and, however rare, when it does occur it is a life-threatening event.2
The clearance of metformin is strongly dependent on kidney function,3 and therefore guidelines still recommend reducing the dose in those with moderate renal insufficiency and recommend considering stopping the medication in those with severe renal insufficiency—the population we were talking about in our article.4 We are aware of changes to the guidelines that have been made by various groups, and in many circumstances we ourselves take an individualized approach, weighing the risks and benefits of continued therapy with the patient and his or her primary care provider. That being said, we did not believe that such nuanced recommendations were appropriate for our article, especially since they are contrary to marketing restrictions for the drug.
In Reply: We appreciate Dr. Imam’s comments regarding using metformin in those with chronic kidney disease.
The US Food and Drug Administration currently lists metformin as contraindicated in those with mild to moderate renal insufficiency, with serum creatinine levels greater than or equal to 1.5 mg/dL in males and greater than or equal to 1.4 mg/dL in females. This contraindication is based on the pharmacokinetics of the medication and, likely, the association of a similar medication, phenformin, with lactic acidosis, which eventually led to its withdrawal from the market. However, lactic acidosis is much less frequent with metformin than with phenformin.1
We agree that metformin is an invaluable medication for diabetes mellitus not requiring insulin. We also agree that lactic acidosis is rare, especially in those with mild renal insufficiency. However, lactic acidosis does occur in patients with chronic kidney disease while on metformin and, however rare, when it does occur it is a life-threatening event.2
The clearance of metformin is strongly dependent on kidney function,3 and therefore guidelines still recommend reducing the dose in those with moderate renal insufficiency and recommend considering stopping the medication in those with severe renal insufficiency—the population we were talking about in our article.4 We are aware of changes to the guidelines that have been made by various groups, and in many circumstances we ourselves take an individualized approach, weighing the risks and benefits of continued therapy with the patient and his or her primary care provider. That being said, we did not believe that such nuanced recommendations were appropriate for our article, especially since they are contrary to marketing restrictions for the drug.
- Bailey CJ, Turner RC. Metformin. N Engl J Med 1996; 334:574–579.
- Lalau JD, Race JM. Lactic acidosis in metformin-treated patients. Prognostic value of arterial lactate levels and plasma metformin concentrations. Drug Saf 1999; 20:377–384.
- Sambol NC, Chiang J, Lin ET, et al. Kidney function and age are both predictors of pharmacokinetics of metformin. J Clin Pharmacol 1995; 35:1094–1102.
- Sakhuja A, Hyland J, Simon JF. Managing advanced chronic kidney disease: a primary care guide. Cleve Clin J Med 2014; 81:289–299.
- Bailey CJ, Turner RC. Metformin. N Engl J Med 1996; 334:574–579.
- Lalau JD, Race JM. Lactic acidosis in metformin-treated patients. Prognostic value of arterial lactate levels and plasma metformin concentrations. Drug Saf 1999; 20:377–384.
- Sambol NC, Chiang J, Lin ET, et al. Kidney function and age are both predictors of pharmacokinetics of metformin. J Clin Pharmacol 1995; 35:1094–1102.
- Sakhuja A, Hyland J, Simon JF. Managing advanced chronic kidney disease: a primary care guide. Cleve Clin J Med 2014; 81:289–299.
Managing advanced chronic kidney disease: A primary care guide
Accountable-care organizations are becoming more prominent in the United States, and therefore health care systems in the near future will be reimbursed on the basis of their ability to care for patient populations rather than individual patients. As a result, primary care physicians will need to be well versed in the care of patients with common chronic diseases such as chronic kidney disease (CKD). By one estimate, patients with CKD constitute 14% of the US population age 20 and older, or more than 31 million people.1
An earlier article in this journal reviewed how to identify patients with CKD and how to interpret the estimated glomerular filtration rate (GFR).2 This article examines the care of patients with advanced CKD, how to manage their health risks, and how to optimize their care by coordinating with nephrologists.
GOALS OF CKD CARE

CKD is defined either as renal damage (which is most commonly manifested by proteinuria, but which may include pathologic changes on biopsy or other markers of damage on serum, urine, or imaging studies), or as a GFR less than 60 mL/min/1.73 m2 for at least 3 months.3 It is divided into five stages (Table 1).
Since most patients with CKD never reach end-stage renal disease, much of their care is aimed at slowing the progression of renal dysfunction and addressing medical issues that arise as a result of CKD. To these ends, it is important to detect CKD early and refer these patients to a nephrology team in a timely manner. Their care can be separated into several important tasks:
- Identify the cause of CKD, if possible; address potentially reversible causes such as obstruction or medication-related causes. If a primarily glomerular process (marked by heavy proteinuria and dysmorphic red blood cells and red blood cell casts in the urine sediment) or interstitial nephritis (manifested by white blood cells in the urine) is suspected, refer to a nephrologist early.
- Provide treatment to correct the specific cause (if one is present) or slow the deterioration of renal function.
- Address cardiovascular risk factors.
- Address metabolic abnormalities related to CKD.
- If the CKD is advanced, educate the patient about end-stage renal disease and its treatment options, and guide the patient through the transition to end-stage renal disease.
WHEN SHOULD A NEPHROLOGIST BE CONSULTED?

The ideal timing of referral to a nephrologist is not well defined and depends on the comfort level of the primary care provider.
Treatments to slow the progression of CKD and decrease cardiovascular risk should begin early in CKD (ie, in stage 3) and can be managed by the primary care provider with guidance from a nephrologist. Patients referred to a nephrologist while in stage 3 have been shown to go longer without CKD progression than those referred in later stages.4 Early referral to a nephrologist has also been associated with a decreased mortality rate.5 The studies that found these trends, however, were limited by the fact that patients with stage 3 CKD are less likely to progress to end-stage renal disease or to die of cardiovascular disease than patients with stage 4 or 5 CKD.
Once stage 4 CKD develops, the nephrologist should take a more active role in the care plan. In this stage, cardiovascular risk rises, and the risk of developing end-stage renal disease rises dramatically.6 With comprehensive care in a CKD clinic, even patients with advanced CKD are more likely to have a stabilization of renal function.7 Kinchen et al8 found that patients referred to a nephrologist within 4 months of starting dialysis had a lower survival rate than those referred earlier. Therefore, if a nephrologist was not involved in the patient’s care prior to stage 4, then a referral must be made.
Recommendation. Patients with stage 3 CKD can be referred for an initial evaluation and development of a treatment plan, but most of the responsibility for their care can remain with the primary care provider. Once stage 4 CKD develops, the nephrologist should assume an increasing role. However, if glomerular disease is suspected, we recommend referral to a nephrologist regardless of the estimated GFR.
ELEVATED CARDIOVASCULAR RISK
Patients with stage 3 CKD are 20 times more likely to die of a cardiovascular event than to reach end-stage renal disease.6 This increased risk does not quite reach the status of a cardiovascular disease risk equivalent, as does diabetes,9,10 but cardiovascular risk reduction should be a primary focus of care for the CKD patient.
The cardiovascular risk in part is attributed to a high prevalence of traditional cardiovascular risk factors, including diabetes mellitus, hypertension, and hyperlipidemia.11,12 About two-thirds of CKD patients have metabolic syndrome, which is a risk factor for cardiovascular disease and is associated with more rapid progression of CKD.13 In addition, renal dysfunction, proteinuria, and hyperphosphatemia are also risk factors for cardiovascular disease.14–19
The risk of death from a cardiovascular event increases as kidney function declines, with reported 5-year death rates of 19.5% in stage 2, 24.3% in stage 3, and 45.7% in stage 4 CKD. However, imbalance between mortality risk and progression to end-stage renal disease may be age-dependent.20 Younger patients (age 45 and younger) are more likely to progress to end-stage renal disease, whereas in older patients (over age 65), the relative risk of dying of cardiovascular disease is higher.
Aggressive lipid management
Hyperlipidemia is a common risk factor for cardiovascular morbidity and mortality in CKD.21 However, until recently, all studies of outcomes of patients treated for hyperlipidemia excluded patients with CKD. Post hoc analyses of these studies 22–27 showed statins to be beneficial in primary and secondary cardiovascular prevention in patents with “normal” serum creatinine values but estimated GFR levels of 50 to 59 mL/min/1.73 m2.
The SHARP trial28 was the first prospective trial to study lipid-lowering therapy in patients with CKD. In this trial, patients with various stages of CKD, including advanced CKD, had fewer major vascular events if they received the combination of low-dose simvastatin (Zocor) and ezetimibe (Zetia). However, the evidence does not suggest that statin therapy slows the progression of CKD.28–31
Recommendation. Manage hyperlipidemia aggressively using statin therapy with or without ezetimibe, with a target low-density lipoprotein cholesterol level below 100 mg/dL.32
Manage other cardiovascular risk factors
Because hypertension and proteinuria are risk factors not only for cardiovascular disease but also for progression of CKD, they are discussed in the section below.
ATTEMPT TO PREVENT WORSENING OF RENAL FUNCTION
Medications to avoid
It is important to review a CKD patient’s medication list—prescription and over-the-counter drugs—to identify any that may contribute to a worsening of renal function. CKD patients need to be informed about avoiding medications such as nonsteroidal anti-inflammatory drugs, proton pump inhibitors, and herbal supplements because they can cause further renal injury. In addition, other medications (eg, metformin) are contraindicated in CKD because of side effects that may occur in CKD.
Patients should be encouraged to discuss any changes in their medications, including over-the-counter products, with their primary care physicians.
Manage hypertension aggressively
Many patients with CKD also have hypertension,33,34 possibly because they have a higher frequency of underlying essential hypertension or because CKD often worsens preexisting hypertension. Moreover, uncontrolled hypertension is associated with a further decline in renal function.35,36
The ACCORD trial37 found no benefit in lowering systolic blood pressure to less than 120 mm Hg compared with less than 140 mm Hg in patients with diabetes mellitus. (The patients in this study did not necessarily have CKD.)
A meta-analysis38 of trials of antihypertensive treatment in patients with CKD found that the optimal target systolic blood pressure for decreasing the progression of CKD was 110 to 129 mm Hg. The relative risk of progression of renal dysfunction was:
- 1.83 (95% confidence interval [CI] 0.97–3.44) at 130 mm to 139 mm Hg, vs
- 3.14 (95% CI 1.64–5.99) at 160 mm Hg or higher.
There is also evidence that blood pressure control can be relaxed as patients age. While the exact age differs among published guidelines, the evidence supports a goal blood pressure of less than 150/90 mm Hg once a patient reaches the age of 70, regardless of CKD or proteinuria.
Recommendation. Current evidence suggests the following blood pressure goals in CKD patients:
- With diabetes mellitus or proteinuria: < 130/80 mm Hg
- Without proteinuria: < 140/90 mm Hg
- Age 70 and older: <150/90 mm Hg.39
Angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) are the preferred antihypertensive drugs in patients with diabetes or proteinuria (see below).
Manage proteinuria
Proteinuria is also associated with progression of CKD. AASK,40 a study that included nondiabetic African American patients whose estimated GFRs were between 20 and 60 mL/min/1.73 m2, showed that higher levels of proteinuria were associated with a higher risk of decline in GFR and a higher risk of end-stage renal disease. Findings were similar to those in studies of other CKD populations.41–43 Proteinuria is also an independent risk factor for cardiovascular disease and death. Multiple large studies16,17,44,45 have found associations between higher levels of albumin excretion and risk of major cardiovascular events, cardiovascular death, and death from any cause in people with and without diabetes.
Reducing proteinuria has been shown to both slow progression of renal dysfunction and reduce the cardiovascular risk.44,45 In a substudy of the IDNT46 in patients with diabetic nephropathy, each 50% reduction in urinary protein excretion was associated with a 56% reduction in risk of progression of CKD. Similar effects have been shown in nondiabetic CKD patients.47
ACE inhibitors and ARBs are the preferred treatments for proteinuria in patients with CKD.48–50 Combination therapy with an ACE inhibitor and an ARB has been used,51–53 with a better response in proteinuria reduction. However, combination therapy with these drugs cannot currently be recommended, as the only prospective study of this regimen to date suggested worse renal and overall outcomes in patients at high cardiovascular risk.54 These drugs may also have renoprotective effects independent of their effects on blood pressure and proteinuria.38 Dietary salt restriction and diuretic therapy can further increase the efficacy of proteinuria reduction by ACE inhibitors or ARBs.55,56
On the other hand, stopping ACE inhibitors or ARBs may be beneficial as the patient nears end-stage renal disease. Ahmed et al57 demonstrated that stopping ACE inhibitors or ARBs in advanced stage 4 CKD (mean estimated GFR 16 mL/min/1.73 m2) was associated with improved GFR and delayed onset of renal replacement therapy. This improvement may be due to regaining the slight decrease in GFR that occurred when these medications were started.
Nondihydropyridine calcium channel blockers such as diltiazem (Cardizem) and verapamil (Calan) have also been shown to be useful for reducing proteinuria,58 whereas dihydropyridine calcium channel blockers such as amlodipine (Norvasc) and nifedipine (Procardia), when used without ACE inhibitors or ARBs, can worsen proteinuria.58,59
Correct metabolic acidosis
The kidneys play an important role in maintaining acid-base balance, keeping the blood from becoming too acidic both by reabsorbing bicarbonate filtered into the urine by the glomerulus and by excreting the daily acid load. Metabolic acidosis can develop when these functions break down at more advanced stages of CKD, most often when the estimated GFR declines to less than 20 mL/min/1.73 m2.
Bicarbonate levels of 22 mmol/L or less have been associated with a higher risk of worsening renal function.60 When such patients were treated with sodium bicarbonate to achieve a serum bicarbonate of at least 23 mmol/L, they had an 80% lower rate of progression to end-stage renal disease without any increase in edema, admission for congestive heart failure, or change in blood pressure.61
Susantitaphong et al62 reviewed six randomized trials of bicarbonate supplementation in CKD and found that it was associated with improved kidney function and a 79% lower rate of progression to end-stage renal disease.
The proposed mechanism behind this benefit lies in the increase in ammonia production that each surviving nephron must undertake to handle the daily acid load. The increased ammonia is thought to play a role in activating the alternative complement pathway,63 causing renal inflammation and injury.
Recommendation. Bicarbonate therapy should be used to maintain serum bicarbonate levels above 22 mmol/L in CKD.64
OTHER ASPECTS OF CKD CARE
Bone mineral disorders
Patients with CKD develop secondary hyperparathyroidism, hyperphosphatemia, and (in advanced CKD) hypocalcemia, all leading to disorders of bone mineral metabolism.
Traditionally, it has been thought that decreased production of 1,25-dihydroxyvitamin D by dysfunctional kidneys leads to decreased suppression of the parathyroid gland and to secondary hyperparathyroidism. The major long-term adverse effect of this is a weakened bone matrix resulting from increased calcium and phosphorus efflux from bones (renal osteodystrophy).
The discovery of fibroblast growth factor 23 (FGF-23) has improved our understanding of the physiology behind disordered bone mineral metabolism in CKD. FGF-23, produced by osteoblasts and osteocytes, acts directly on the kidney to increase renal phosphate excretion. It also suppresses 1,25-dihydroxyvitamin D levels by inhibiting 1-alpha-hydroxylase,65 and it stimulates parathyroid hormone secretion. FGF-23 levels rise much earlier in CKD than do parathyroid hormone levels, suggesting that abnormalities in phosphorus balance and FGF-23 may be the earliest pathophysiologic changes.66
The initial treatment of bone mineral disorders is to some extent guided by laboratory values. Phosphate levels higher than 3.5 or 4 mg/dL and elevated FGF-23 levels have been associated with increased mortality rates in CKD patients.18,19,67–69 All patients should also have their 1,25-dihydroxyvitamin D level checked and supplemented if deficient. In many patients with early stage 3 CKD, this may correct secondary hyperparathyroidism.70
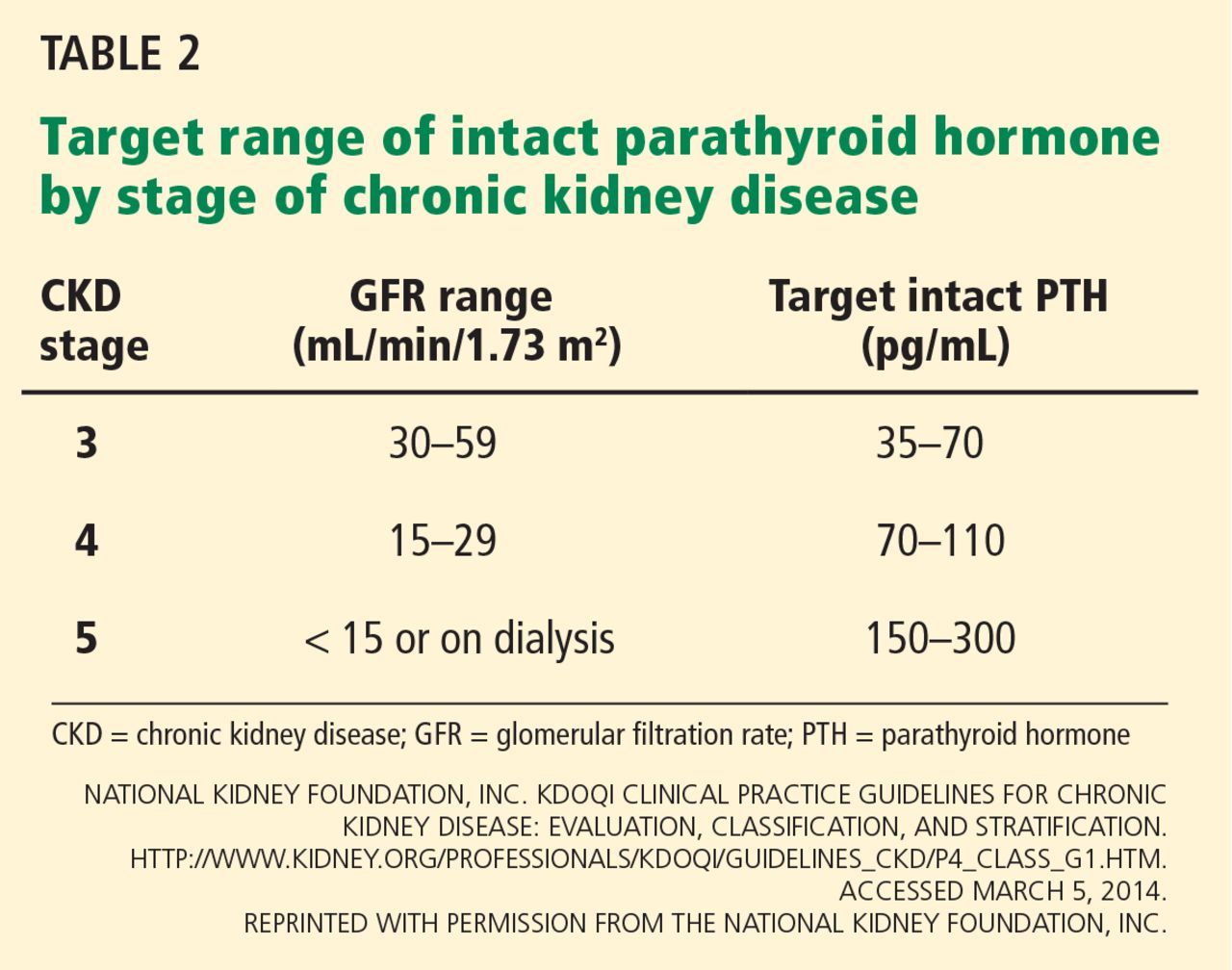
Serum phosphorus levels should be kept in the normal range in stage 3 and 4 CKD,71 either by restricting dietary phosphorus intake (< 800 or < 1,000 mg/day) or by using a phosphate binder, which is taken with meals to prevent phosphorus absorption from the gastrointestinal tract. Current US recommendations are to allow graded increases in parathyroid hormone based on the stage of CKD (Table 2).71 However, these targets are still an area of uncertainty, with some guidelines suggesting that wider variations in parathyroid hormone can be allowed, so there may be wider variation in clinical practice in this area.72 If the serum phosphorus level is in the goal range but parathyroid hormone levels are still high, an activated vitamin D analogue such as calcitriol is recommended, although with the emerging role of FGF-23, some experts also call for early use of a phosphate binder in this group.
The treatment of bone mineral disorders in CKD is fairly complex, and we recommend that it be done by or with the close direction of a nephrologist.
Recommendations on bone disorders
- Check levels of calcium, phosphorus, 25-hydroxyvitamin D, and parathyroid hormone in all patients whose estimated GFR is less than 60 mL/min/1.73 m2, with frequency of measurements based on the stage of CKD.71
- Replace vitamin D if deficient.
- Treat elevated phosphorus levels with a protein-restricted diet (nutrition referral) and a phosphate binder.
- Treat elevated hyperparathyroid hormone levels with a vitamin D analogue once phosphorus levels have been controlled.
- Refer patients with an elevated phosphorus or parathyroid hormone level to a nephrology service for consultation before initiating medical therapy.
Anemia is common, treatment controversial
The treatment of anemia attributed to CKD has been a topic of controversy over the past decade, and we recommend that it be done with the guidance of a nephrologist.
Anemia is common in CKD, and declining kidney function is an independent predictor of anemia.73 Anemia is a risk factor for left ventricular hypertrophy, cardiovascular disease,74 and death in CKD.75
The anemia of CKD is attributed to relative erythropoietin deficiency and bone marrow resistance to erythropoietin, but this is a diagnosis of exclusion, and other causes of anemia must be ruled out. Iron deficiency is a common cause of anemia in CKD, and treatment of iron deficiency may correct anemia in more than one-third of these patients.76,77
Erythropoiesis-stimulating agents such as epoetin alfa (Procrit) and darbepoetin (Aranesp) are used to treat renal anemia. However, the target hemoglobin level has been a subject of debate. Three prospective trials78–80 found no benefit in raising the hemoglobin level to normal ranges using these agents, and several found an association with higher rates of stroke and venous thrombosis. The US Food and Drug Administration suggests that the only role for these agents in CKD is to avoid the need for transfusions. They should not be used to normalize the hemoglobin level. The target, although not explicitly specified, is suggested to be around 10 g/dL.81
PREPARE FOR END-STAGE RENAL DISEASE
Discuss the options
Because the risk of developing end-stage renal disease rises dramatically once CKD reaches stage 4, all such patients should have a discussion about renal replacement therapy. They should be educated about their options for treatment (hemodialysis, peritoneal dialysis, and transplantation, as well as not proceeding with renal replacement therapy), often in a formal class. They should then be actively engaged in the decision about how to proceed. Survival and quality of life should be discussed, particularly with patients who are over age 80, who are severely ill, or who are living in a nursing facility, as these groups get limited survival benefit from starting dialysis, and quality of life may actually decrease with dialysis.82,83
The Renal Physicians Association has created clinical practice guidelines for shared decision-making, consisting of 10 practice recommendations that outline a systematic approach to patients needing renal replacement therapy.84
Consider preemptive kidney transplantation
Any patient thought to be a suitable candidate for renal transplantation should be referred to a transplantation center for evaluation. Studies have shown that kidney transplantation offers a survival advantage compared with chronic dialysis and should preferably be done preemptively, ie, before dialysis is required.85–90 Therefore, patients with estimated GFRs in the low 20s should be referred for a transplantation evaluation.
If a living donor is available, the transplantation team usually waits to perform the procedure until the patient is closer to needing dialysis, often when the estimated GFR is around 15 to 16 mL/min/1.73 m2. If no living donor is available, the patient can earn time on the deceased-donor waiting list once his or her estimated GFR falls to below 20 mL/min/1.7 m2.
Plan for dialysis access

Patients starting hemodialysis first need to undergo a procedure to provide access to the blood. The three options are an arteriovenous fistula, an arteriovenous graft, and a central venous catheter (Figure 1).
An arteriovenous fistula is the best option, being the most durable, followed by a graft and then a catheter.91 Arteriovenous fistulas also have the lowest rates of infection,92 thrombosis,93 and intervention to maintain patency.93
The fistula is created by ligating a vein draining an extremity, most often the nondominant arm, and anastomosing the vein to an artery. The higher arterial pressure causes the vein to dilate and thicken (“arterialize”), thus making it able to withstand repeated cannulation necessary for hemodialysis.
An arteriovenous fistula typically takes 1 to 3 months to “mature” to the point where it can be used,94,95 and, depending on the patient and experience of the vascular surgeon, a significant number may never mature. Thus, it is important to discuss hemodialysis access before the patient reaches end-stage renal disease so that he or she can be referred to a vascular surgeon early, when the estimated GFR is about 20 mL/min/1.73 m2.
An arteriovenous graft. Not all patients have suitable vessels for creation of an arteriovenous fistula. In such patients, an arteriovenous graft, typically made of polytetrafluoroethylene, is the next best option. The graft is typically ready to use in 2 weeks and thus does not require as much advance planning. Grafts tend to narrow more often than fistulas and require more procedures to keep them patent.
A central venous catheter is most often inserted into the internal jugular vein and tunneled under the skin to exit in an area covered by the patient’s shirt.
Tunneled dialysis catheters are associated with higher rates of infection, thrombosis, and overall mortality and are therefore the least preferred choice. They are reserved for patients who have not had advance planning for end-stage renal disease, who do not have acceptable vessels for an arteriovenous fistula or graft, or who have refused surgical access.
Protect the fistula arm. It is recommended that venipuncture, intravenous lines, and blood pressure measurements be avoided in the nondominant upper arm of patients with stage 4 and 5 CKD to protect those veins for the potential creation of an arteriovenous fistula.96 For the same reason, peripherally inserted central catheter lines and subclavian catheters should be avoided in these patients. If an arteriovenous fistula has already been placed, this arm must be protected from such procedures at all times.
Studies have shown that late referral to a nephrologist is associated with a lower incidence of starting dialysis with a permanent vascular access.97,98
If the patient wishes to start peritoneal dialysis, the peritoneal dialysis catheter can usually be used 2 weeks after being inserted.
Starting dialysis
The appropriate time for starting dialysis remains controversial, especially in elderly patients with multiple comorbid conditions.
The IDEAL study99 found no benefit in starting dialysis at a GFR of 10 to 14 mL/min compared with 5 to 7 mL/min. Thus, there is no single estimated GFR at which dialysis should be started. Rather, the development of early uremic symptoms and the patient’s quality of life should guide this decision.82,83,99–101
Hemodialysis involves three sessions per week, each taking about 4 hours. Evidence suggests that longer sessions or more sessions per week may offer benefits, especially in terms of blood pressure, volume, and dietary management. This has led to an increase in the popularity of home and in-center nocturnal hemodialysis programs across the United States.
Peritoneal dialysis?
Peritoneal dialysis is an excellent choice for patients who are motivated, can care for themselves at home, and have a support system available to assist them if needed. It allows for daily dialysis, less fluid restriction, and less dietary restriction, and it gives the patient an opportunity to stay independent. It also spares the veins in the arms, which may be needed for vascular access later in life if hemodialysis is needed.
Recommendation. We recommend that peritoneal dialysis be offered to any suitable patient who is approaching end-stage renal disease.
A COMPREHENSIVE, COLLABORATIVE APPROACH

Chronic kidney disease is a multisystem disorder, and its management requires a comprehensive approach (Table 3). Early detection and interventions are key to reducing cardiovascular events and progression to kidney failure.
Early referral to a nephrologist and team collaboration between the primary care provider, the nephrologist, and other health care providers are essential. Early in the course of CKD, it may be appropriate for a nephrologist to evaluate the patient and recommend a set of treatment goals. Follow-up may be infrequent or unnecessary.
As CKD progresses, especially as the patient reaches an estimated GFR of 30 mL/min/1.73 m2, the nephrologist will take a more active role in the patient’s care and medical decision-making. In some circumstances, it may even be appropriate for the nephrologist to be the patient’s source of primary care, with the primary care provider as a consultant.
Caring for patients with CKD includes not only strategies to preserve renal function and prolong survival, but also making critical decisions about starting dialysis and about the need for transplantation. Early involvement of a nephrologist and early preparation for end-stage renal disease with preemptive transplantation and arteriovenous fistula placement are associated with better patient outcomes. Key to this is collaboration between the primary care provider and the nephrologist, with levels of responsibility for patient care that adapt to the patient’s degree of renal dysfunction and other comorbidities. Such strategies to select patients for timely nephrology referral may help improve outcomes in this vulnerable population.
- United States Renal Data System (USRDS). Identification and care of patients with CKD. http://www.usrds.org/2012/pdf/v1_ch2_12.pdf. Accessed March 5, 2014.
- Simon J, Amde M, Poggio ED. Interpreting the estimated glomerular filtration rate in primary care: benefits and pitfalls. Cleve Clin J Med 2011; 78:189–195.
- National Kidney Foundation, Inc. KDOQI Clinical Practice Guidelines for Chronic Kidney Disease: Evaluation, Classification, and Stratification. http://www.kidney.org/professionals/kdoqi/guidelines_ckd/p4_class_g1.htm. Accessed March 5, 2014.
- Orlando LA, Owen WF, Matchar DB. Relationship between nephrologist care and progression of chronic kidney disease. N C Med J 2007; 68:9–16.
- Tseng CL, Kern EF, Miller DR, et al. Survival benefit of nephrologic care in patients with diabetes mellitus and chronic kidney disease. Arch Intern Med 2008; 168:55–62.
- Keith DS, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med 2004; 164:659–663.
- Serrano A, Huang J, Ghossein C, et al. Stabilization of glomerular filtration rate in advanced chronic kidney disease: a two-year follow-up of a cohort of chronic kidney disease patients stages 4 and 5. Adv Chronic Kidney Dis 2007; 14:105–112.
- Kinchen KS, Sadler J, Fink N, et al. The timing of specialist evaluation in chronic kidney disease and mortality. Ann Intern Med 2002; 137:479–486.
- Tonelli M, Muntner P, Lloyd A, et al; Alberta Kidney Disease Network. Risk of coronary events in people with chronic kidney disease compared with those with diabetes: a population-level cohort study. Lancet 2012; 380:807–814.
- Wattanakit K, Coresh J, Muntner P, Marsh J, Folsom AR. Cardiovascular risk among adults with chronic kidney disease, with or without prior myocardial infarction. J Am Coll Cardiol 2006; 48:1183–1189.
- Foley RN, Wang C, Collins AJ. Cardiovascular risk factor profiles and kidney function stage in the US general population: the NHANES III study. Mayo Clin Proc 2005; 80:1270–1277.
- Muntner P, He J, Astor BC, Folsom AR, Coresh J. Traditional and nontraditional risk factors predict coronary heart disease in chronic kidney disease: results from the Atherosclerosis Risk in Communities Study. J Am Soc Nephrol 2005; 16:529–538.
- Navaneethan SD, Schold JD, Kirwan JP, et al. Metabolic syndrome, ESRD, and death in CKD. Clin J Am Soc Nephrol 2013; 8:945–952.
- Foley RN, Murray AM, Li S, et al. Chronic kidney disease and the risk for cardiovascular disease, renal replacement, and death in the United States Medicare population, 1998 to 1999. J Am Soc Nephrol 2005; 16:489–495.
- Weiner DE, Tighiouart H, Amin MG, et al. Chronic kidney disease as a risk factor for cardiovascular disease and all-cause mortality: a pooled analysis of community-based studies. J Am Soc Nephrol 2004; 15:1307–1315.
- Gerstein HC, Mann JF, Yi Q, et al; HOPE Study Investigators. Albuminuria and risk of cardiovascular events, death, and heart failure in diabetic and nondiabetic individuals. JAMA 2001; 286:421–426.
- Hillege HL, Fidler V, Diercks GF, et al; Prevention of Renal and Vascular End Stage Disease (PREVEND) Study Group. Urinary albumin excretion predicts cardiovascular and noncardiovascular mortality in general population. Circulation 2002; 106:1777–1782.
- Foley RN, Collins AJ, Ishani A, Kalra PA. Calcium-phosphate levels and cardiovascular disease in community-dwelling adults: the Atherosclerosis Risk in Communities (ARIC) Study. Am Heart J 2008; 156:556–563.
- Tonelli M, Sacks F, Pfeffer M, Gao Z, Curhan G; Cholesterol And Recurrent Events Trial Investigators. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 2005; 112:2627–2633.
- Menon V, Wang X, Sarnak MJ, et al. Long-term outcomes in nondiabetic chronic kidney disease. Kidney Int 2008; 73:1310–1315.
- Kasiske BL. Hyperlipidemia in patients with chronic renal disease. Am J Kidney Dis 1998; 32(suppl 3):S142–S156.
- Kendrick J, Shlipak MG, Targher G, Cook T, Lindenfeld J, Chonchol M. Effect of lovastatin on primary prevention of cardiovascular events in mild CKD and kidney function loss: a post hoc analysis of the Air Force/Texas Coronary Atherosclerosis Prevention Study. Am J Kidney Dis 2010; 55:42–49.
- Colhoun HM, Betteridge DJ, Durrington PN, et al; CARDS Investigators. Effects of atorvastatin on kidney outcomes and cardiovascular disease in patients with diabetes: an analysis from the Collaborative Atorvastatin Diabetes Study (CARDS). Am J Kidney Dis 2009; 54:810–819.
- Koren MJ, Davidson MH, Wilson DJ, Fayyad RS, Zuckerman A, Reed DP; ALLIANCE Investigators. Focused atorvastatin therapy in managed-care patients with coronary heart disease and CKD. Am J Kidney Dis 2009; 53:741–750.
- Fellström BC, Jardine AG, Schmieder RE, et al; AURORA Study Group. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N Engl J Med 2009; 360:1395–1407.
- Chonchol M, Cook T, Kjekshus J, Pedersen TR, Lindenfeld J. Simvastatin for secondary prevention of all-cause mortality and major coronary events in patients with mild chronic renal insufficiency. Am J Kidney Dis 2007; 49:373–382.
- Ridker PM, MacFadyen J, Cressman M, Glynn RJ. Efficacy of rosuvastatin among men and women with moderate chronic kidney disease and elevated high-sensitivity C-reactive protein: a secondary analysis from the JUPITER (Justification for the Use of Statins in Prevention-an Intervention Trial Evaluating Rosuvastatin) trial. J Am Coll Cardiol 2010; 55:1266–1273.
- Baigent C, Landray MJ, Reith C, et al; SHARP Investigators. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet 2011; 377:2181–2192.
- Shepherd J, Kastelein JJ, Bittner V, et al; Treating to New Targets Investigators. Effect of intensive lipid lowering with atorvastatin on renal function in patients with coronary heart disease: the Treating to New Targets (TNT) study. Clin J Am Soc Nephrol 2007; 2:1131–1139.
- Tonelli M, Isles C, Craven T, et al. Effect of pravastatin on rate of kidney function loss in people with or at risk for coronary disease. Circulation 2005; 112:171–178.
- Palmer SC, Craig JC, Navaneethan SD, Tonelli M, Pellegrini F, Strippoli GF. Benefits and harms of statin therapy for persons with chronic kidney disease: a systematic review and meta-analysis. Ann Intern Med 2012; 157:263–275.
- National Kidney Foundation, Inc. KDOQI Clinical Practice Guidelines for Managing Dyslipidemias in Chronic Kidney Disease. http://www.kidney.org/professionals/kdoqi/guidelines_lipids/. Accessed March 5, 2014.
- Buckalew VM, Berg RL, Wang SR, Porush JG, Rauch S, Schulman G. Prevalence of hypertension in 1,795 subjects with chronic renal disease: the modification of diet in renal disease study baseline cohort. Modification of Diet in Renal Disease Study Group. Am J Kidney Dis 1996; 28:811–821.
- Coresh J, Wei GL, McQuillan G, et al. Prevalence of high blood pressure and elevated serum creatinine level in the United States: findings from the third National Health and Nutrition Examination Survey (1988–1994). Arch Intern Med 2001; 161:1207–1216.
- Klag MJ, Whelton PK, Randall BL, et al. Blood pressure and end-stage renal disease in men. N Engl J Med 1996; 334:13–18.
- Locatelli F, Marcelli D, Comelli M, et al. Proteinuria and blood pressure as causal components of progression to end-stage renal failure. Northern Italian Cooperative Study Group. Nephrol Dial Transplant 1996; 11:461–467.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- Jafar TH, Stark PC, Schmid CH, et al. Progression of chronic kidney disease: the role of blood pressure control, proteinuria, and angiotensin-converting enzyme inhibition: a patient-level meta-analysis. Ann Intern Med 2003; 139:244–252.
- Khosla N, Bakris G. Lessons learned from recent hypertension trials about kidney disease. Clin J Am Soc Nephrol 2006; 1:229–235.
- Norris KC, Greene T, Kopple J, et al. Baseline predictors of renal disease progression in the African American Study of Hypertension and Kidney Disease. J Am Soc Nephrol 2006; 17:2928–2936.
- Keane WF, Brenner BM, de Zeeuw D, et al; RENAAL Study Investigators. The risk of developing end-stage renal disease in patients with type 2 diabetes and nephropathy: the RENAAL study. Kidney Int 2003; 63:1499–1507.
- Ruggenenti P, Perna A, Mosconi L, et al. Proteinuria predicts end-stage renal failure in non-diabetic chronic nephropathies. The “Gruppo Italiano di Studi Epidemiologici in Nefrologia” (GISEN). Kidney Int Suppl 1997; 63:S54–S57.
- de Goeij MC, Liem M, de Jager DJ, et al; PREPARE-1 Study Group. Proteinuria as a risk marker for the progression of chronic kidney disease in patients on predialysis care and the role of angiotensin-converting enzyme inhibitor/angiotensin II receptor blocker treatment. Nephron Clin Pract 2012; 121:c73–c82.
- de Zeeuw D, Remuzzi G, Parving HH, et al. Albuminuria, a therapeutic target for cardiovascular protection in type 2 diabetic patients with nephropathy. Circulation 2004; 110:921–927.
- Ibsen H, Olsen MH, Wachtell K, et al. Reduction in albuminuria translates to reduction in cardiovascular events in hypertensive patients: losartan intervention for endpoint reduction in hypertension study. Hypertension 2005; 45:198–202.
- Atkins RC, Briganti EM, Lewis JB, et al. Proteinuria reduction and progression to renal failure in patients with type 2 diabetes mellitus and overt nephropathy. Am J Kidney Dis 2005; 45:281–287.
- Jafar TH, Stark PC, Schmid CH, et al; AIPRD Study Group; Angiotensin-Converting Enzyme Inhibition and Progression of Renal Disease. Proteinuria as a modifiable risk factor for the progression of non-diabetic renal disease. Kidney Int 2001; 60:1131–1140.
- ACE Inhibitors in Diabetic Nephropathy Trialist Group. Should all patients with type 1 diabetes mellitus and microalbuminuria receive angiotensin-converting enzyme inhibitors? A meta-analysis of individual patient data. Ann Intern Med 2001; 134:370–379.
- Casas JP, Chua W, Loukogeorgakis S, et al. Effect of inhibitors of the renin-angiotensin system and other antihypertensive drugs on renal outcomes: systematic review and meta-analysis. Lancet 2005; 366:2026–2033.
- Strippoli GF, Craig M, Deeks JJ, Schena FP, Craig JC. Effects of angiotensin converting enzyme inhibitors and angiotensin II receptor antagonists on mortality and renal outcomes in diabetic nephropathy: systematic review. BMJ 2004; 329:828.
- MacKinnon M, Shurraw S, Akbari A, Knoll GA, Jaffey J, Clark HD. Combination therapy with an angiotensin receptor blocker and an ACE inhibitor in proteinuric renal disease: a systematic review of the efficacy and safety data. Am J Kidney Dis 2006; 48:8–20.
- Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of mono-therapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med 2008; 148:30–48.
- Ruggenenti P, Perticucci E, Cravedi P, et al. Role of remission clinics in the longitudinal treatment of CKD. J Am Soc Nephrol 2008; 19:1213–1224.
- Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Esnault VL, Ekhlas A, Delcroix C, Moutel MG, Nguyen JM. Diuretic and enhanced sodium restriction results in improved antiproteinuric response to RAS blocking agents. J Am Soc Nephrol 2005; 16:474–481.
- Vogt L, Waanders F, Boomsma F, de Zeeuw D, Navis G. Effects of dietary sodium and hydrochlorothiazide on the antiproteinuric efficacy of losartan. J Am Soc Nephrol 2008; 19:999–1007.
- Ahmed AK, Kamath NS, El Kossi M, El Nahas AM. The impact of stopping inhibitors of the renin-angiotensin system in patients with advanced chronic kidney disease. Nephrol Dial Transplant 2010; 25:3977–3982.
- Bakris GL, Weir MR, Secic M, Campbell B, Weis-McNulty A. Differential effects of calcium antagonist subclasses on markers of nephropathy progression. Kidney Int 2004; 65:1991–2002.
- Kloke HJ, Wetzels JF, Koene RA, Huysmans FT. Effects of low-dose nifedipine on urinary protein excretion rate in patients with renal disease. Nephrol Dial Transplant 1998; 13:646–650.
- Shah SN, Abramowitz M, Hostetter TH, Melamed ML. Serum bicarbonate levels and the progression of kidney disease: a cohort study. Am J Kidney Dis 2009; 54:270–277.
- de Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol 2009; 20:2075–2084.
- Susantitaphong P, Sewaralthahab K, Balk EM, Jaber BL, Madias NE. Short- and long-term effects of alkali therapy in chronic kidney disease: a systematic review. Am J Nephrol 2012; 35:540–547.
- Nath KA, Hostetter MK, Hostetter TH. Ammonia-complement interaction in the pathogenesis of progressive renal injury. Kidney Int Suppl 1989; 27:S52–S54.
- Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am J Kidney Dis 2000; 35(suppl 2):S1–S140.
- Shimada T, Yamazaki Y, Takahashi M, et al. Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am J Physiol Renal Physiol 2005; 289:F1088–F1095.
- Hasegawa H, Nagano N, Urakawa I, et al. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int 2010; 78:975–980.
- de Boer IH, Rue TC, Kestenbaum B. Serum phosphorus concentrations in the third National Health and Nutrition Examination Survey (NHANES III). Am J Kidney Dis 2009; 53:399–407.
- Kendrick J, Cheung AK, Kaufman JS, et al; HOST Investigators. FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J Am Soc Nephrol 2011; 22:1913–1922.
- Palmer SC, Hayen A, Macaskill P, et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. JAMA. 2011; 305:1119–1127.
- Kooienga L, Fried L, Scragg R, Kendrick J, Smits G, Chonchol M. The effect of combined calcium and vitamin D3 supplementation on serum intact parathyroid hormone in moderate CKD. Am J Kidney Dis 2009; 53:408–416.
- National Kidney Foundation, Inc. KDOQI Clinical Practice Guidelines for Bone Metabolism and Disease in Chronic Kidney Disease. www.kidney.org/professionals/kdoqi/guidelines_bone/guide1.htm#table15. Accessed March 5, 2014.
- Kidney International. KDIGO Clinical Practice Guideline for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). http://kdigo.org/home/mineral-bone-disorder. Accessed March 5, 2014.
- Kazmi WH, Kausz AT, Khan S, et al. Anemia: an early complication of chronic renal insufficiency. Am J Kidney Dis 2001; 38:803–812.
- Sarnak MJ, Tighiouart H, Manjunath G, et al. Anemia as a risk factor for cardiovascular disease in the Atherosclerosis Risk in Communities (ARIC) study. J Am Coll Cardiol 2002; 40:27–33.
- Thorp ML, Johnson ES, Yang X, Petrik AF, Platt R, Smith DH. Effect of anaemia on mortality, cardiovascular hospitalizations and end-stage renal disease among patients with chronic kidney disease. Nephrology (Carlton) 2009; 14:240–246.
- Mircescu G, Gârneata L, Capusa C, Ursea N. Intravenous iron supplementation for the treatment of anaemia in pre-dialyzed chronic renal failure patients. Nephrol Dial Transplant 2006; 21:120–124.
- Silverberg DS, Iaina A, Peer G, et al. Intravenous iron supplementation for the treatment of the anemia of moderate to severe chronic renal failure patients not receiving dialysis. Am J Kidney Dis 1996; 27:234–238.
- Singh AK, Szczech L, Tang KL, et al; CHOIR Investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- Pfeffer MA, Burdmann EA, Chen CY, et al; TREAT Investigators. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 2009; 361:2019–2032.
- US Food and Drug Administration (FDA). FDA Drug Safety Communication: modified dosing recommendations to improve the safe use of erythropoiesis-stimulating agents (ESAs) in chronic kidney disease. http://www.fda.gov/drugs/drugsafety/ucm259639.htm. Accessed March 5, 2014.
- Kurella M, Covinsky KE, Collins AJ, Chertow GM. Octogenarians and nonagenarians starting dialysis in the United States. Ann Intern Med 2007; 146:177–183.
- Kurella Tamura M, Covinsky KE, Chertow GM, Yaffe K, Landefeld CS, McCulloch CE. Functional status of elderly adults before and after initiation of dialysis. N Engl J Med 2009; 361:1539–1547.
- Renal Physicians Association. Clinical Practice Guideline. Shared Decision-Making in the Appropriate Initiation of and Withdrawal from Dialysis. 2nd ed.
- Vollmer WM, Wahl PW, Blagg CR. Survival with dialysis and transplantation in patients with end-stage renal disease. N Engl J Med 1983; 308:1553–1558.
- Port FK, Wolfe RA, Mauger EA, Berling DP, Jiang K. Comparison of survival probabilities for dialysis patients vs cadaveric renal transplant recipients. JAMA 1993; 270:1339–1343.
- Wolfe RA, Ashby VB, Milford EL, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med 1999; 341:1725–1730.
- Cosio FG, Alamir A, Yim S, et al. Patient survival after renal transplantation: I. The impact of dialysis pre-transplant. Kidney Int 1998; 53:767–772.
- Meier-Kriesche HU, Port FK, Ojo AO, et al. Effect of waiting time on renal transplant outcome. Kidney Int 2000; 58:1311–1317.
- Mange KC, Joffe MM, Feldman HI. Effect of the use or nonuse of long-term dialysis on the subsequent survival of renal transplants from living donors. N Engl J Med 2001; 344:726–731.
- Dhingra RK, Young EW, Hulbert-Shearon TE, Leavey SF, Port FK. Type of vascular access and mortality in US hemodialysis patients. Kidney Int 2001; 60:1443–1451.
- Nassar GM, Ayus JC. Infectious complications of the hemodialysis access. Kidney Int 2001; 60:1–13.
- Perera GB, Mueller MP, Kubaska SM, Wilson SE, Lawrence PF, Fujitani RM. Superiority of autogenous arteriovenous hemodialysis access: maintenance of function with fewer secondary interventions. Ann Vasc Surg 2004; 18:66–73.
- Basile C, Casucci F, Lomonte C. Timing of first cannulation of arteriovenous fistula: time matters, but there is also something else. Nephrol Dial Transplant 2005; 20:1519–1520.
- Biuckians A, Scott EC, Meier GH, Panneton JM, Glickman MH. The natural history of autologous fistulas as first-time dialysis access in the KDOQI era. J Vasc Surg 2008; 47:415–421.
- National Kidney Foundation, Inc. KDOQI Clinical Practice Guidelines for Vascular Access. http://www.kidney.org/professionals/KDOQI/guideline_upHD_PD_VA/va_guide1.htm. Accessed March 5, 2014.
- Arora P, Obrador GT, Ruthazer R, et al. Prevalence, predictors, and consequences of late nephrology referral at a tertiary care center. J Am Soc Nephrol 1999; 10:1281–1286.
- Gøransson LG, Bergrem H. Consequences of late referral of patients with end-stage renal disease. J Intern Med 2001; 250:154–159.
- Cooper BA, Branley P, Bulfone L, et al. A randomized, controlled trial of early versus late initiation of dialysis. N Engl J Med 2010; 363:609–619.
- Carson RC, Juszczak M, Davenport A, Burns A. Is maximum conservative management an equivalent treatment option to dialysis for elderly patients with significant comorbid disease? Clin J Am Soc Nephrol 2009; 4:1611–1619.
- Murtagh FE, Marsh JE, Donohoe P, Ekbal NJ, Sheerin NS, Harris FE. Dialysis or not? A comparative survival study of patients over 75 years with chronic kidney disease stage 5. Nephrol Dial Transplant 2007; 22:1955–1962.
Accountable-care organizations are becoming more prominent in the United States, and therefore health care systems in the near future will be reimbursed on the basis of their ability to care for patient populations rather than individual patients. As a result, primary care physicians will need to be well versed in the care of patients with common chronic diseases such as chronic kidney disease (CKD). By one estimate, patients with CKD constitute 14% of the US population age 20 and older, or more than 31 million people.1
An earlier article in this journal reviewed how to identify patients with CKD and how to interpret the estimated glomerular filtration rate (GFR).2 This article examines the care of patients with advanced CKD, how to manage their health risks, and how to optimize their care by coordinating with nephrologists.
GOALS OF CKD CARE
CKD is defined either as renal damage (which is most commonly manifested by proteinuria, but which may include pathologic changes on biopsy or other markers of damage on serum, urine, or imaging studies), or as a GFR less than 60 mL/min/1.73 m2 for at least 3 months.3 It is divided into five stages (Table 1).
Since most patients with CKD never reach end-stage renal disease, much of their care is aimed at slowing the progression of renal dysfunction and addressing medical issues that arise as a result of CKD. To these ends, it is important to detect CKD early and refer these patients to a nephrology team in a timely manner. Their care can be separated into several important tasks:
- Identify the cause of CKD, if possible; address potentially reversible causes such as obstruction or medication-related causes. If a primarily glomerular process (marked by heavy proteinuria and dysmorphic red blood cells and red blood cell casts in the urine sediment) or interstitial nephritis (manifested by white blood cells in the urine) is suspected, refer to a nephrologist early.
- Provide treatment to correct the specific cause (if one is present) or slow the deterioration of renal function.
- Address cardiovascular risk factors.
- Address metabolic abnormalities related to CKD.
- If the CKD is advanced, educate the patient about end-stage renal disease and its treatment options, and guide the patient through the transition to end-stage renal disease.
WHEN SHOULD A NEPHROLOGIST BE CONSULTED?
The ideal timing of referral to a nephrologist is not well defined and depends on the comfort level of the primary care provider.
Treatments to slow the progression of CKD and decrease cardiovascular risk should begin early in CKD (ie, in stage 3) and can be managed by the primary care provider with guidance from a nephrologist. Patients referred to a nephrologist while in stage 3 have been shown to go longer without CKD progression than those referred in later stages.4 Early referral to a nephrologist has also been associated with a decreased mortality rate.5 The studies that found these trends, however, were limited by the fact that patients with stage 3 CKD are less likely to progress to end-stage renal disease or to die of cardiovascular disease than patients with stage 4 or 5 CKD.
Once stage 4 CKD develops, the nephrologist should take a more active role in the care plan. In this stage, cardiovascular risk rises, and the risk of developing end-stage renal disease rises dramatically.6 With comprehensive care in a CKD clinic, even patients with advanced CKD are more likely to have a stabilization of renal function.7 Kinchen et al8 found that patients referred to a nephrologist within 4 months of starting dialysis had a lower survival rate than those referred earlier. Therefore, if a nephrologist was not involved in the patient’s care prior to stage 4, then a referral must be made.
Recommendation. Patients with stage 3 CKD can be referred for an initial evaluation and development of a treatment plan, but most of the responsibility for their care can remain with the primary care provider. Once stage 4 CKD develops, the nephrologist should assume an increasing role. However, if glomerular disease is suspected, we recommend referral to a nephrologist regardless of the estimated GFR.
ELEVATED CARDIOVASCULAR RISK
Patients with stage 3 CKD are 20 times more likely to die of a cardiovascular event than to reach end-stage renal disease.6 This increased risk does not quite reach the status of a cardiovascular disease risk equivalent, as does diabetes,9,10 but cardiovascular risk reduction should be a primary focus of care for the CKD patient.
The cardiovascular risk in part is attributed to a high prevalence of traditional cardiovascular risk factors, including diabetes mellitus, hypertension, and hyperlipidemia.11,12 About two-thirds of CKD patients have metabolic syndrome, which is a risk factor for cardiovascular disease and is associated with more rapid progression of CKD.13 In addition, renal dysfunction, proteinuria, and hyperphosphatemia are also risk factors for cardiovascular disease.14–19
The risk of death from a cardiovascular event increases as kidney function declines, with reported 5-year death rates of 19.5% in stage 2, 24.3% in stage 3, and 45.7% in stage 4 CKD. However, imbalance between mortality risk and progression to end-stage renal disease may be age-dependent.20 Younger patients (age 45 and younger) are more likely to progress to end-stage renal disease, whereas in older patients (over age 65), the relative risk of dying of cardiovascular disease is higher.
Aggressive lipid management
Hyperlipidemia is a common risk factor for cardiovascular morbidity and mortality in CKD.21 However, until recently, all studies of outcomes of patients treated for hyperlipidemia excluded patients with CKD. Post hoc analyses of these studies 22–27 showed statins to be beneficial in primary and secondary cardiovascular prevention in patents with “normal” serum creatinine values but estimated GFR levels of 50 to 59 mL/min/1.73 m2.
The SHARP trial28 was the first prospective trial to study lipid-lowering therapy in patients with CKD. In this trial, patients with various stages of CKD, including advanced CKD, had fewer major vascular events if they received the combination of low-dose simvastatin (Zocor) and ezetimibe (Zetia). However, the evidence does not suggest that statin therapy slows the progression of CKD.28–31
Recommendation. Manage hyperlipidemia aggressively using statin therapy with or without ezetimibe, with a target low-density lipoprotein cholesterol level below 100 mg/dL.32
Manage other cardiovascular risk factors
Because hypertension and proteinuria are risk factors not only for cardiovascular disease but also for progression of CKD, they are discussed in the section below.
ATTEMPT TO PREVENT WORSENING OF RENAL FUNCTION
Medications to avoid
It is important to review a CKD patient’s medication list—prescription and over-the-counter drugs—to identify any that may contribute to a worsening of renal function. CKD patients need to be informed about avoiding medications such as nonsteroidal anti-inflammatory drugs, proton pump inhibitors, and herbal supplements because they can cause further renal injury. In addition, other medications (eg, metformin) are contraindicated in CKD because of side effects that may occur in CKD.
Patients should be encouraged to discuss any changes in their medications, including over-the-counter products, with their primary care physicians.
Manage hypertension aggressively
Many patients with CKD also have hypertension,33,34 possibly because they have a higher frequency of underlying essential hypertension or because CKD often worsens preexisting hypertension. Moreover, uncontrolled hypertension is associated with a further decline in renal function.35,36
The ACCORD trial37 found no benefit in lowering systolic blood pressure to less than 120 mm Hg compared with less than 140 mm Hg in patients with diabetes mellitus. (The patients in this study did not necessarily have CKD.)
A meta-analysis38 of trials of antihypertensive treatment in patients with CKD found that the optimal target systolic blood pressure for decreasing the progression of CKD was 110 to 129 mm Hg. The relative risk of progression of renal dysfunction was:
- 1.83 (95% confidence interval [CI] 0.97–3.44) at 130 mm to 139 mm Hg, vs
- 3.14 (95% CI 1.64–5.99) at 160 mm Hg or higher.
There is also evidence that blood pressure control can be relaxed as patients age. While the exact age differs among published guidelines, the evidence supports a goal blood pressure of less than 150/90 mm Hg once a patient reaches the age of 70, regardless of CKD or proteinuria.
Recommendation. Current evidence suggests the following blood pressure goals in CKD patients:
- With diabetes mellitus or proteinuria: < 130/80 mm Hg
- Without proteinuria: < 140/90 mm Hg
- Age 70 and older: <150/90 mm Hg.39
Angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) are the preferred antihypertensive drugs in patients with diabetes or proteinuria (see below).
Manage proteinuria
Proteinuria is also associated with progression of CKD. AASK,40 a study that included nondiabetic African American patients whose estimated GFRs were between 20 and 60 mL/min/1.73 m2, showed that higher levels of proteinuria were associated with a higher risk of decline in GFR and a higher risk of end-stage renal disease. Findings were similar to those in studies of other CKD populations.41–43 Proteinuria is also an independent risk factor for cardiovascular disease and death. Multiple large studies16,17,44,45 have found associations between higher levels of albumin excretion and risk of major cardiovascular events, cardiovascular death, and death from any cause in people with and without diabetes.
Reducing proteinuria has been shown to both slow progression of renal dysfunction and reduce the cardiovascular risk.44,45 In a substudy of the IDNT46 in patients with diabetic nephropathy, each 50% reduction in urinary protein excretion was associated with a 56% reduction in risk of progression of CKD. Similar effects have been shown in nondiabetic CKD patients.47
ACE inhibitors and ARBs are the preferred treatments for proteinuria in patients with CKD.48–50 Combination therapy with an ACE inhibitor and an ARB has been used,51–53 with a better response in proteinuria reduction. However, combination therapy with these drugs cannot currently be recommended, as the only prospective study of this regimen to date suggested worse renal and overall outcomes in patients at high cardiovascular risk.54 These drugs may also have renoprotective effects independent of their effects on blood pressure and proteinuria.38 Dietary salt restriction and diuretic therapy can further increase the efficacy of proteinuria reduction by ACE inhibitors or ARBs.55,56
On the other hand, stopping ACE inhibitors or ARBs may be beneficial as the patient nears end-stage renal disease. Ahmed et al57 demonstrated that stopping ACE inhibitors or ARBs in advanced stage 4 CKD (mean estimated GFR 16 mL/min/1.73 m2) was associated with improved GFR and delayed onset of renal replacement therapy. This improvement may be due to regaining the slight decrease in GFR that occurred when these medications were started.
Nondihydropyridine calcium channel blockers such as diltiazem (Cardizem) and verapamil (Calan) have also been shown to be useful for reducing proteinuria,58 whereas dihydropyridine calcium channel blockers such as amlodipine (Norvasc) and nifedipine (Procardia), when used without ACE inhibitors or ARBs, can worsen proteinuria.58,59
Correct metabolic acidosis
The kidneys play an important role in maintaining acid-base balance, keeping the blood from becoming too acidic both by reabsorbing bicarbonate filtered into the urine by the glomerulus and by excreting the daily acid load. Metabolic acidosis can develop when these functions break down at more advanced stages of CKD, most often when the estimated GFR declines to less than 20 mL/min/1.73 m2.
Bicarbonate levels of 22 mmol/L or less have been associated with a higher risk of worsening renal function.60 When such patients were treated with sodium bicarbonate to achieve a serum bicarbonate of at least 23 mmol/L, they had an 80% lower rate of progression to end-stage renal disease without any increase in edema, admission for congestive heart failure, or change in blood pressure.61
Susantitaphong et al62 reviewed six randomized trials of bicarbonate supplementation in CKD and found that it was associated with improved kidney function and a 79% lower rate of progression to end-stage renal disease.
The proposed mechanism behind this benefit lies in the increase in ammonia production that each surviving nephron must undertake to handle the daily acid load. The increased ammonia is thought to play a role in activating the alternative complement pathway,63 causing renal inflammation and injury.
Recommendation. Bicarbonate therapy should be used to maintain serum bicarbonate levels above 22 mmol/L in CKD.64
OTHER ASPECTS OF CKD CARE
Bone mineral disorders
Patients with CKD develop secondary hyperparathyroidism, hyperphosphatemia, and (in advanced CKD) hypocalcemia, all leading to disorders of bone mineral metabolism.
Traditionally, it has been thought that decreased production of 1,25-dihydroxyvitamin D by dysfunctional kidneys leads to decreased suppression of the parathyroid gland and to secondary hyperparathyroidism. The major long-term adverse effect of this is a weakened bone matrix resulting from increased calcium and phosphorus efflux from bones (renal osteodystrophy).
The discovery of fibroblast growth factor 23 (FGF-23) has improved our understanding of the physiology behind disordered bone mineral metabolism in CKD. FGF-23, produced by osteoblasts and osteocytes, acts directly on the kidney to increase renal phosphate excretion. It also suppresses 1,25-dihydroxyvitamin D levels by inhibiting 1-alpha-hydroxylase,65 and it stimulates parathyroid hormone secretion. FGF-23 levels rise much earlier in CKD than do parathyroid hormone levels, suggesting that abnormalities in phosphorus balance and FGF-23 may be the earliest pathophysiologic changes.66
The initial treatment of bone mineral disorders is to some extent guided by laboratory values. Phosphate levels higher than 3.5 or 4 mg/dL and elevated FGF-23 levels have been associated with increased mortality rates in CKD patients.18,19,67–69 All patients should also have their 1,25-dihydroxyvitamin D level checked and supplemented if deficient. In many patients with early stage 3 CKD, this may correct secondary hyperparathyroidism.70
Serum phosphorus levels should be kept in the normal range in stage 3 and 4 CKD,71 either by restricting dietary phosphorus intake (< 800 or < 1,000 mg/day) or by using a phosphate binder, which is taken with meals to prevent phosphorus absorption from the gastrointestinal tract. Current US recommendations are to allow graded increases in parathyroid hormone based on the stage of CKD (Table 2).71 However, these targets are still an area of uncertainty, with some guidelines suggesting that wider variations in parathyroid hormone can be allowed, so there may be wider variation in clinical practice in this area.72 If the serum phosphorus level is in the goal range but parathyroid hormone levels are still high, an activated vitamin D analogue such as calcitriol is recommended, although with the emerging role of FGF-23, some experts also call for early use of a phosphate binder in this group.
The treatment of bone mineral disorders in CKD is fairly complex, and we recommend that it be done by or with the close direction of a nephrologist.
Recommendations on bone disorders
- Check levels of calcium, phosphorus, 25-hydroxyvitamin D, and parathyroid hormone in all patients whose estimated GFR is less than 60 mL/min/1.73 m2, with frequency of measurements based on the stage of CKD.71
- Replace vitamin D if deficient.
- Treat elevated phosphorus levels with a protein-restricted diet (nutrition referral) and a phosphate binder.
- Treat elevated hyperparathyroid hormone levels with a vitamin D analogue once phosphorus levels have been controlled.
- Refer patients with an elevated phosphorus or parathyroid hormone level to a nephrology service for consultation before initiating medical therapy.
Anemia is common, treatment controversial
The treatment of anemia attributed to CKD has been a topic of controversy over the past decade, and we recommend that it be done with the guidance of a nephrologist.
Anemia is common in CKD, and declining kidney function is an independent predictor of anemia.73 Anemia is a risk factor for left ventricular hypertrophy, cardiovascular disease,74 and death in CKD.75
The anemia of CKD is attributed to relative erythropoietin deficiency and bone marrow resistance to erythropoietin, but this is a diagnosis of exclusion, and other causes of anemia must be ruled out. Iron deficiency is a common cause of anemia in CKD, and treatment of iron deficiency may correct anemia in more than one-third of these patients.76,77
Erythropoiesis-stimulating agents such as epoetin alfa (Procrit) and darbepoetin (Aranesp) are used to treat renal anemia. However, the target hemoglobin level has been a subject of debate. Three prospective trials78–80 found no benefit in raising the hemoglobin level to normal ranges using these agents, and several found an association with higher rates of stroke and venous thrombosis. The US Food and Drug Administration suggests that the only role for these agents in CKD is to avoid the need for transfusions. They should not be used to normalize the hemoglobin level. The target, although not explicitly specified, is suggested to be around 10 g/dL.81
PREPARE FOR END-STAGE RENAL DISEASE
Discuss the options
Because the risk of developing end-stage renal disease rises dramatically once CKD reaches stage 4, all such patients should have a discussion about renal replacement therapy. They should be educated about their options for treatment (hemodialysis, peritoneal dialysis, and transplantation, as well as not proceeding with renal replacement therapy), often in a formal class. They should then be actively engaged in the decision about how to proceed. Survival and quality of life should be discussed, particularly with patients who are over age 80, who are severely ill, or who are living in a nursing facility, as these groups get limited survival benefit from starting dialysis, and quality of life may actually decrease with dialysis.82,83
The Renal Physicians Association has created clinical practice guidelines for shared decision-making, consisting of 10 practice recommendations that outline a systematic approach to patients needing renal replacement therapy.84
Consider preemptive kidney transplantation
Any patient thought to be a suitable candidate for renal transplantation should be referred to a transplantation center for evaluation. Studies have shown that kidney transplantation offers a survival advantage compared with chronic dialysis and should preferably be done preemptively, ie, before dialysis is required.85–90 Therefore, patients with estimated GFRs in the low 20s should be referred for a transplantation evaluation.
If a living donor is available, the transplantation team usually waits to perform the procedure until the patient is closer to needing dialysis, often when the estimated GFR is around 15 to 16 mL/min/1.73 m2. If no living donor is available, the patient can earn time on the deceased-donor waiting list once his or her estimated GFR falls to below 20 mL/min/1.7 m2.
Plan for dialysis access
Patients starting hemodialysis first need to undergo a procedure to provide access to the blood. The three options are an arteriovenous fistula, an arteriovenous graft, and a central venous catheter (Figure 1).
An arteriovenous fistula is the best option, being the most durable, followed by a graft and then a catheter.91 Arteriovenous fistulas also have the lowest rates of infection,92 thrombosis,93 and intervention to maintain patency.93
The fistula is created by ligating a vein draining an extremity, most often the nondominant arm, and anastomosing the vein to an artery. The higher arterial pressure causes the vein to dilate and thicken (“arterialize”), thus making it able to withstand repeated cannulation necessary for hemodialysis.
An arteriovenous fistula typically takes 1 to 3 months to “mature” to the point where it can be used,94,95 and, depending on the patient and experience of the vascular surgeon, a significant number may never mature. Thus, it is important to discuss hemodialysis access before the patient reaches end-stage renal disease so that he or she can be referred to a vascular surgeon early, when the estimated GFR is about 20 mL/min/1.73 m2.
An arteriovenous graft. Not all patients have suitable vessels for creation of an arteriovenous fistula. In such patients, an arteriovenous graft, typically made of polytetrafluoroethylene, is the next best option. The graft is typically ready to use in 2 weeks and thus does not require as much advance planning. Grafts tend to narrow more often than fistulas and require more procedures to keep them patent.
A central venous catheter is most often inserted into the internal jugular vein and tunneled under the skin to exit in an area covered by the patient’s shirt.
Tunneled dialysis catheters are associated with higher rates of infection, thrombosis, and overall mortality and are therefore the least preferred choice. They are reserved for patients who have not had advance planning for end-stage renal disease, who do not have acceptable vessels for an arteriovenous fistula or graft, or who have refused surgical access.
Protect the fistula arm. It is recommended that venipuncture, intravenous lines, and blood pressure measurements be avoided in the nondominant upper arm of patients with stage 4 and 5 CKD to protect those veins for the potential creation of an arteriovenous fistula.96 For the same reason, peripherally inserted central catheter lines and subclavian catheters should be avoided in these patients. If an arteriovenous fistula has already been placed, this arm must be protected from such procedures at all times.
Studies have shown that late referral to a nephrologist is associated with a lower incidence of starting dialysis with a permanent vascular access.97,98
If the patient wishes to start peritoneal dialysis, the peritoneal dialysis catheter can usually be used 2 weeks after being inserted.
Starting dialysis
The appropriate time for starting dialysis remains controversial, especially in elderly patients with multiple comorbid conditions.
The IDEAL study99 found no benefit in starting dialysis at a GFR of 10 to 14 mL/min compared with 5 to 7 mL/min. Thus, there is no single estimated GFR at which dialysis should be started. Rather, the development of early uremic symptoms and the patient’s quality of life should guide this decision.82,83,99–101
Hemodialysis involves three sessions per week, each taking about 4 hours. Evidence suggests that longer sessions or more sessions per week may offer benefits, especially in terms of blood pressure, volume, and dietary management. This has led to an increase in the popularity of home and in-center nocturnal hemodialysis programs across the United States.
Peritoneal dialysis?
Peritoneal dialysis is an excellent choice for patients who are motivated, can care for themselves at home, and have a support system available to assist them if needed. It allows for daily dialysis, less fluid restriction, and less dietary restriction, and it gives the patient an opportunity to stay independent. It also spares the veins in the arms, which may be needed for vascular access later in life if hemodialysis is needed.
Recommendation. We recommend that peritoneal dialysis be offered to any suitable patient who is approaching end-stage renal disease.
A COMPREHENSIVE, COLLABORATIVE APPROACH
Chronic kidney disease is a multisystem disorder, and its management requires a comprehensive approach (Table 3). Early detection and interventions are key to reducing cardiovascular events and progression to kidney failure.
Early referral to a nephrologist and team collaboration between the primary care provider, the nephrologist, and other health care providers are essential. Early in the course of CKD, it may be appropriate for a nephrologist to evaluate the patient and recommend a set of treatment goals. Follow-up may be infrequent or unnecessary.
As CKD progresses, especially as the patient reaches an estimated GFR of 30 mL/min/1.73 m2, the nephrologist will take a more active role in the patient’s care and medical decision-making. In some circumstances, it may even be appropriate for the nephrologist to be the patient’s source of primary care, with the primary care provider as a consultant.
Caring for patients with CKD includes not only strategies to preserve renal function and prolong survival, but also making critical decisions about starting dialysis and about the need for transplantation. Early involvement of a nephrologist and early preparation for end-stage renal disease with preemptive transplantation and arteriovenous fistula placement are associated with better patient outcomes. Key to this is collaboration between the primary care provider and the nephrologist, with levels of responsibility for patient care that adapt to the patient’s degree of renal dysfunction and other comorbidities. Such strategies to select patients for timely nephrology referral may help improve outcomes in this vulnerable population.
Accountable-care organizations are becoming more prominent in the United States, and therefore health care systems in the near future will be reimbursed on the basis of their ability to care for patient populations rather than individual patients. As a result, primary care physicians will need to be well versed in the care of patients with common chronic diseases such as chronic kidney disease (CKD). By one estimate, patients with CKD constitute 14% of the US population age 20 and older, or more than 31 million people.1
An earlier article in this journal reviewed how to identify patients with CKD and how to interpret the estimated glomerular filtration rate (GFR).2 This article examines the care of patients with advanced CKD, how to manage their health risks, and how to optimize their care by coordinating with nephrologists.
GOALS OF CKD CARE
CKD is defined either as renal damage (which is most commonly manifested by proteinuria, but which may include pathologic changes on biopsy or other markers of damage on serum, urine, or imaging studies), or as a GFR less than 60 mL/min/1.73 m2 for at least 3 months.3 It is divided into five stages (Table 1).
Since most patients with CKD never reach end-stage renal disease, much of their care is aimed at slowing the progression of renal dysfunction and addressing medical issues that arise as a result of CKD. To these ends, it is important to detect CKD early and refer these patients to a nephrology team in a timely manner. Their care can be separated into several important tasks:
- Identify the cause of CKD, if possible; address potentially reversible causes such as obstruction or medication-related causes. If a primarily glomerular process (marked by heavy proteinuria and dysmorphic red blood cells and red blood cell casts in the urine sediment) or interstitial nephritis (manifested by white blood cells in the urine) is suspected, refer to a nephrologist early.
- Provide treatment to correct the specific cause (if one is present) or slow the deterioration of renal function.
- Address cardiovascular risk factors.
- Address metabolic abnormalities related to CKD.
- If the CKD is advanced, educate the patient about end-stage renal disease and its treatment options, and guide the patient through the transition to end-stage renal disease.
WHEN SHOULD A NEPHROLOGIST BE CONSULTED?
The ideal timing of referral to a nephrologist is not well defined and depends on the comfort level of the primary care provider.
Treatments to slow the progression of CKD and decrease cardiovascular risk should begin early in CKD (ie, in stage 3) and can be managed by the primary care provider with guidance from a nephrologist. Patients referred to a nephrologist while in stage 3 have been shown to go longer without CKD progression than those referred in later stages.4 Early referral to a nephrologist has also been associated with a decreased mortality rate.5 The studies that found these trends, however, were limited by the fact that patients with stage 3 CKD are less likely to progress to end-stage renal disease or to die of cardiovascular disease than patients with stage 4 or 5 CKD.
Once stage 4 CKD develops, the nephrologist should take a more active role in the care plan. In this stage, cardiovascular risk rises, and the risk of developing end-stage renal disease rises dramatically.6 With comprehensive care in a CKD clinic, even patients with advanced CKD are more likely to have a stabilization of renal function.7 Kinchen et al8 found that patients referred to a nephrologist within 4 months of starting dialysis had a lower survival rate than those referred earlier. Therefore, if a nephrologist was not involved in the patient’s care prior to stage 4, then a referral must be made.
Recommendation. Patients with stage 3 CKD can be referred for an initial evaluation and development of a treatment plan, but most of the responsibility for their care can remain with the primary care provider. Once stage 4 CKD develops, the nephrologist should assume an increasing role. However, if glomerular disease is suspected, we recommend referral to a nephrologist regardless of the estimated GFR.
ELEVATED CARDIOVASCULAR RISK
Patients with stage 3 CKD are 20 times more likely to die of a cardiovascular event than to reach end-stage renal disease.6 This increased risk does not quite reach the status of a cardiovascular disease risk equivalent, as does diabetes,9,10 but cardiovascular risk reduction should be a primary focus of care for the CKD patient.
The cardiovascular risk in part is attributed to a high prevalence of traditional cardiovascular risk factors, including diabetes mellitus, hypertension, and hyperlipidemia.11,12 About two-thirds of CKD patients have metabolic syndrome, which is a risk factor for cardiovascular disease and is associated with more rapid progression of CKD.13 In addition, renal dysfunction, proteinuria, and hyperphosphatemia are also risk factors for cardiovascular disease.14–19
The risk of death from a cardiovascular event increases as kidney function declines, with reported 5-year death rates of 19.5% in stage 2, 24.3% in stage 3, and 45.7% in stage 4 CKD. However, imbalance between mortality risk and progression to end-stage renal disease may be age-dependent.20 Younger patients (age 45 and younger) are more likely to progress to end-stage renal disease, whereas in older patients (over age 65), the relative risk of dying of cardiovascular disease is higher.
Aggressive lipid management
Hyperlipidemia is a common risk factor for cardiovascular morbidity and mortality in CKD.21 However, until recently, all studies of outcomes of patients treated for hyperlipidemia excluded patients with CKD. Post hoc analyses of these studies 22–27 showed statins to be beneficial in primary and secondary cardiovascular prevention in patents with “normal” serum creatinine values but estimated GFR levels of 50 to 59 mL/min/1.73 m2.
The SHARP trial28 was the first prospective trial to study lipid-lowering therapy in patients with CKD. In this trial, patients with various stages of CKD, including advanced CKD, had fewer major vascular events if they received the combination of low-dose simvastatin (Zocor) and ezetimibe (Zetia). However, the evidence does not suggest that statin therapy slows the progression of CKD.28–31
Recommendation. Manage hyperlipidemia aggressively using statin therapy with or without ezetimibe, with a target low-density lipoprotein cholesterol level below 100 mg/dL.32
Manage other cardiovascular risk factors
Because hypertension and proteinuria are risk factors not only for cardiovascular disease but also for progression of CKD, they are discussed in the section below.
ATTEMPT TO PREVENT WORSENING OF RENAL FUNCTION
Medications to avoid
It is important to review a CKD patient’s medication list—prescription and over-the-counter drugs—to identify any that may contribute to a worsening of renal function. CKD patients need to be informed about avoiding medications such as nonsteroidal anti-inflammatory drugs, proton pump inhibitors, and herbal supplements because they can cause further renal injury. In addition, other medications (eg, metformin) are contraindicated in CKD because of side effects that may occur in CKD.
Patients should be encouraged to discuss any changes in their medications, including over-the-counter products, with their primary care physicians.
Manage hypertension aggressively
Many patients with CKD also have hypertension,33,34 possibly because they have a higher frequency of underlying essential hypertension or because CKD often worsens preexisting hypertension. Moreover, uncontrolled hypertension is associated with a further decline in renal function.35,36
The ACCORD trial37 found no benefit in lowering systolic blood pressure to less than 120 mm Hg compared with less than 140 mm Hg in patients with diabetes mellitus. (The patients in this study did not necessarily have CKD.)
A meta-analysis38 of trials of antihypertensive treatment in patients with CKD found that the optimal target systolic blood pressure for decreasing the progression of CKD was 110 to 129 mm Hg. The relative risk of progression of renal dysfunction was:
- 1.83 (95% confidence interval [CI] 0.97–3.44) at 130 mm to 139 mm Hg, vs
- 3.14 (95% CI 1.64–5.99) at 160 mm Hg or higher.
There is also evidence that blood pressure control can be relaxed as patients age. While the exact age differs among published guidelines, the evidence supports a goal blood pressure of less than 150/90 mm Hg once a patient reaches the age of 70, regardless of CKD or proteinuria.
Recommendation. Current evidence suggests the following blood pressure goals in CKD patients:
- With diabetes mellitus or proteinuria: < 130/80 mm Hg
- Without proteinuria: < 140/90 mm Hg
- Age 70 and older: <150/90 mm Hg.39
Angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) are the preferred antihypertensive drugs in patients with diabetes or proteinuria (see below).
Manage proteinuria
Proteinuria is also associated with progression of CKD. AASK,40 a study that included nondiabetic African American patients whose estimated GFRs were between 20 and 60 mL/min/1.73 m2, showed that higher levels of proteinuria were associated with a higher risk of decline in GFR and a higher risk of end-stage renal disease. Findings were similar to those in studies of other CKD populations.41–43 Proteinuria is also an independent risk factor for cardiovascular disease and death. Multiple large studies16,17,44,45 have found associations between higher levels of albumin excretion and risk of major cardiovascular events, cardiovascular death, and death from any cause in people with and without diabetes.
Reducing proteinuria has been shown to both slow progression of renal dysfunction and reduce the cardiovascular risk.44,45 In a substudy of the IDNT46 in patients with diabetic nephropathy, each 50% reduction in urinary protein excretion was associated with a 56% reduction in risk of progression of CKD. Similar effects have been shown in nondiabetic CKD patients.47
ACE inhibitors and ARBs are the preferred treatments for proteinuria in patients with CKD.48–50 Combination therapy with an ACE inhibitor and an ARB has been used,51–53 with a better response in proteinuria reduction. However, combination therapy with these drugs cannot currently be recommended, as the only prospective study of this regimen to date suggested worse renal and overall outcomes in patients at high cardiovascular risk.54 These drugs may also have renoprotective effects independent of their effects on blood pressure and proteinuria.38 Dietary salt restriction and diuretic therapy can further increase the efficacy of proteinuria reduction by ACE inhibitors or ARBs.55,56
On the other hand, stopping ACE inhibitors or ARBs may be beneficial as the patient nears end-stage renal disease. Ahmed et al57 demonstrated that stopping ACE inhibitors or ARBs in advanced stage 4 CKD (mean estimated GFR 16 mL/min/1.73 m2) was associated with improved GFR and delayed onset of renal replacement therapy. This improvement may be due to regaining the slight decrease in GFR that occurred when these medications were started.
Nondihydropyridine calcium channel blockers such as diltiazem (Cardizem) and verapamil (Calan) have also been shown to be useful for reducing proteinuria,58 whereas dihydropyridine calcium channel blockers such as amlodipine (Norvasc) and nifedipine (Procardia), when used without ACE inhibitors or ARBs, can worsen proteinuria.58,59
Correct metabolic acidosis
The kidneys play an important role in maintaining acid-base balance, keeping the blood from becoming too acidic both by reabsorbing bicarbonate filtered into the urine by the glomerulus and by excreting the daily acid load. Metabolic acidosis can develop when these functions break down at more advanced stages of CKD, most often when the estimated GFR declines to less than 20 mL/min/1.73 m2.
Bicarbonate levels of 22 mmol/L or less have been associated with a higher risk of worsening renal function.60 When such patients were treated with sodium bicarbonate to achieve a serum bicarbonate of at least 23 mmol/L, they had an 80% lower rate of progression to end-stage renal disease without any increase in edema, admission for congestive heart failure, or change in blood pressure.61
Susantitaphong et al62 reviewed six randomized trials of bicarbonate supplementation in CKD and found that it was associated with improved kidney function and a 79% lower rate of progression to end-stage renal disease.
The proposed mechanism behind this benefit lies in the increase in ammonia production that each surviving nephron must undertake to handle the daily acid load. The increased ammonia is thought to play a role in activating the alternative complement pathway,63 causing renal inflammation and injury.
Recommendation. Bicarbonate therapy should be used to maintain serum bicarbonate levels above 22 mmol/L in CKD.64
OTHER ASPECTS OF CKD CARE
Bone mineral disorders
Patients with CKD develop secondary hyperparathyroidism, hyperphosphatemia, and (in advanced CKD) hypocalcemia, all leading to disorders of bone mineral metabolism.
Traditionally, it has been thought that decreased production of 1,25-dihydroxyvitamin D by dysfunctional kidneys leads to decreased suppression of the parathyroid gland and to secondary hyperparathyroidism. The major long-term adverse effect of this is a weakened bone matrix resulting from increased calcium and phosphorus efflux from bones (renal osteodystrophy).
The discovery of fibroblast growth factor 23 (FGF-23) has improved our understanding of the physiology behind disordered bone mineral metabolism in CKD. FGF-23, produced by osteoblasts and osteocytes, acts directly on the kidney to increase renal phosphate excretion. It also suppresses 1,25-dihydroxyvitamin D levels by inhibiting 1-alpha-hydroxylase,65 and it stimulates parathyroid hormone secretion. FGF-23 levels rise much earlier in CKD than do parathyroid hormone levels, suggesting that abnormalities in phosphorus balance and FGF-23 may be the earliest pathophysiologic changes.66
The initial treatment of bone mineral disorders is to some extent guided by laboratory values. Phosphate levels higher than 3.5 or 4 mg/dL and elevated FGF-23 levels have been associated with increased mortality rates in CKD patients.18,19,67–69 All patients should also have their 1,25-dihydroxyvitamin D level checked and supplemented if deficient. In many patients with early stage 3 CKD, this may correct secondary hyperparathyroidism.70
Serum phosphorus levels should be kept in the normal range in stage 3 and 4 CKD,71 either by restricting dietary phosphorus intake (< 800 or < 1,000 mg/day) or by using a phosphate binder, which is taken with meals to prevent phosphorus absorption from the gastrointestinal tract. Current US recommendations are to allow graded increases in parathyroid hormone based on the stage of CKD (Table 2).71 However, these targets are still an area of uncertainty, with some guidelines suggesting that wider variations in parathyroid hormone can be allowed, so there may be wider variation in clinical practice in this area.72 If the serum phosphorus level is in the goal range but parathyroid hormone levels are still high, an activated vitamin D analogue such as calcitriol is recommended, although with the emerging role of FGF-23, some experts also call for early use of a phosphate binder in this group.
The treatment of bone mineral disorders in CKD is fairly complex, and we recommend that it be done by or with the close direction of a nephrologist.
Recommendations on bone disorders
- Check levels of calcium, phosphorus, 25-hydroxyvitamin D, and parathyroid hormone in all patients whose estimated GFR is less than 60 mL/min/1.73 m2, with frequency of measurements based on the stage of CKD.71
- Replace vitamin D if deficient.
- Treat elevated phosphorus levels with a protein-restricted diet (nutrition referral) and a phosphate binder.
- Treat elevated hyperparathyroid hormone levels with a vitamin D analogue once phosphorus levels have been controlled.
- Refer patients with an elevated phosphorus or parathyroid hormone level to a nephrology service for consultation before initiating medical therapy.
Anemia is common, treatment controversial
The treatment of anemia attributed to CKD has been a topic of controversy over the past decade, and we recommend that it be done with the guidance of a nephrologist.
Anemia is common in CKD, and declining kidney function is an independent predictor of anemia.73 Anemia is a risk factor for left ventricular hypertrophy, cardiovascular disease,74 and death in CKD.75
The anemia of CKD is attributed to relative erythropoietin deficiency and bone marrow resistance to erythropoietin, but this is a diagnosis of exclusion, and other causes of anemia must be ruled out. Iron deficiency is a common cause of anemia in CKD, and treatment of iron deficiency may correct anemia in more than one-third of these patients.76,77
Erythropoiesis-stimulating agents such as epoetin alfa (Procrit) and darbepoetin (Aranesp) are used to treat renal anemia. However, the target hemoglobin level has been a subject of debate. Three prospective trials78–80 found no benefit in raising the hemoglobin level to normal ranges using these agents, and several found an association with higher rates of stroke and venous thrombosis. The US Food and Drug Administration suggests that the only role for these agents in CKD is to avoid the need for transfusions. They should not be used to normalize the hemoglobin level. The target, although not explicitly specified, is suggested to be around 10 g/dL.81
PREPARE FOR END-STAGE RENAL DISEASE
Discuss the options
Because the risk of developing end-stage renal disease rises dramatically once CKD reaches stage 4, all such patients should have a discussion about renal replacement therapy. They should be educated about their options for treatment (hemodialysis, peritoneal dialysis, and transplantation, as well as not proceeding with renal replacement therapy), often in a formal class. They should then be actively engaged in the decision about how to proceed. Survival and quality of life should be discussed, particularly with patients who are over age 80, who are severely ill, or who are living in a nursing facility, as these groups get limited survival benefit from starting dialysis, and quality of life may actually decrease with dialysis.82,83
The Renal Physicians Association has created clinical practice guidelines for shared decision-making, consisting of 10 practice recommendations that outline a systematic approach to patients needing renal replacement therapy.84
Consider preemptive kidney transplantation
Any patient thought to be a suitable candidate for renal transplantation should be referred to a transplantation center for evaluation. Studies have shown that kidney transplantation offers a survival advantage compared with chronic dialysis and should preferably be done preemptively, ie, before dialysis is required.85–90 Therefore, patients with estimated GFRs in the low 20s should be referred for a transplantation evaluation.
If a living donor is available, the transplantation team usually waits to perform the procedure until the patient is closer to needing dialysis, often when the estimated GFR is around 15 to 16 mL/min/1.73 m2. If no living donor is available, the patient can earn time on the deceased-donor waiting list once his or her estimated GFR falls to below 20 mL/min/1.7 m2.
Plan for dialysis access
Patients starting hemodialysis first need to undergo a procedure to provide access to the blood. The three options are an arteriovenous fistula, an arteriovenous graft, and a central venous catheter (Figure 1).
An arteriovenous fistula is the best option, being the most durable, followed by a graft and then a catheter.91 Arteriovenous fistulas also have the lowest rates of infection,92 thrombosis,93 and intervention to maintain patency.93
The fistula is created by ligating a vein draining an extremity, most often the nondominant arm, and anastomosing the vein to an artery. The higher arterial pressure causes the vein to dilate and thicken (“arterialize”), thus making it able to withstand repeated cannulation necessary for hemodialysis.
An arteriovenous fistula typically takes 1 to 3 months to “mature” to the point where it can be used,94,95 and, depending on the patient and experience of the vascular surgeon, a significant number may never mature. Thus, it is important to discuss hemodialysis access before the patient reaches end-stage renal disease so that he or she can be referred to a vascular surgeon early, when the estimated GFR is about 20 mL/min/1.73 m2.
An arteriovenous graft. Not all patients have suitable vessels for creation of an arteriovenous fistula. In such patients, an arteriovenous graft, typically made of polytetrafluoroethylene, is the next best option. The graft is typically ready to use in 2 weeks and thus does not require as much advance planning. Grafts tend to narrow more often than fistulas and require more procedures to keep them patent.
A central venous catheter is most often inserted into the internal jugular vein and tunneled under the skin to exit in an area covered by the patient’s shirt.
Tunneled dialysis catheters are associated with higher rates of infection, thrombosis, and overall mortality and are therefore the least preferred choice. They are reserved for patients who have not had advance planning for end-stage renal disease, who do not have acceptable vessels for an arteriovenous fistula or graft, or who have refused surgical access.
Protect the fistula arm. It is recommended that venipuncture, intravenous lines, and blood pressure measurements be avoided in the nondominant upper arm of patients with stage 4 and 5 CKD to protect those veins for the potential creation of an arteriovenous fistula.96 For the same reason, peripherally inserted central catheter lines and subclavian catheters should be avoided in these patients. If an arteriovenous fistula has already been placed, this arm must be protected from such procedures at all times.
Studies have shown that late referral to a nephrologist is associated with a lower incidence of starting dialysis with a permanent vascular access.97,98
If the patient wishes to start peritoneal dialysis, the peritoneal dialysis catheter can usually be used 2 weeks after being inserted.
Starting dialysis
The appropriate time for starting dialysis remains controversial, especially in elderly patients with multiple comorbid conditions.
The IDEAL study99 found no benefit in starting dialysis at a GFR of 10 to 14 mL/min compared with 5 to 7 mL/min. Thus, there is no single estimated GFR at which dialysis should be started. Rather, the development of early uremic symptoms and the patient’s quality of life should guide this decision.82,83,99–101
Hemodialysis involves three sessions per week, each taking about 4 hours. Evidence suggests that longer sessions or more sessions per week may offer benefits, especially in terms of blood pressure, volume, and dietary management. This has led to an increase in the popularity of home and in-center nocturnal hemodialysis programs across the United States.
Peritoneal dialysis?
Peritoneal dialysis is an excellent choice for patients who are motivated, can care for themselves at home, and have a support system available to assist them if needed. It allows for daily dialysis, less fluid restriction, and less dietary restriction, and it gives the patient an opportunity to stay independent. It also spares the veins in the arms, which may be needed for vascular access later in life if hemodialysis is needed.
Recommendation. We recommend that peritoneal dialysis be offered to any suitable patient who is approaching end-stage renal disease.
A COMPREHENSIVE, COLLABORATIVE APPROACH
Chronic kidney disease is a multisystem disorder, and its management requires a comprehensive approach (Table 3). Early detection and interventions are key to reducing cardiovascular events and progression to kidney failure.
Early referral to a nephrologist and team collaboration between the primary care provider, the nephrologist, and other health care providers are essential. Early in the course of CKD, it may be appropriate for a nephrologist to evaluate the patient and recommend a set of treatment goals. Follow-up may be infrequent or unnecessary.
As CKD progresses, especially as the patient reaches an estimated GFR of 30 mL/min/1.73 m2, the nephrologist will take a more active role in the patient’s care and medical decision-making. In some circumstances, it may even be appropriate for the nephrologist to be the patient’s source of primary care, with the primary care provider as a consultant.
Caring for patients with CKD includes not only strategies to preserve renal function and prolong survival, but also making critical decisions about starting dialysis and about the need for transplantation. Early involvement of a nephrologist and early preparation for end-stage renal disease with preemptive transplantation and arteriovenous fistula placement are associated with better patient outcomes. Key to this is collaboration between the primary care provider and the nephrologist, with levels of responsibility for patient care that adapt to the patient’s degree of renal dysfunction and other comorbidities. Such strategies to select patients for timely nephrology referral may help improve outcomes in this vulnerable population.
- United States Renal Data System (USRDS). Identification and care of patients with CKD. http://www.usrds.org/2012/pdf/v1_ch2_12.pdf. Accessed March 5, 2014.
- Simon J, Amde M, Poggio ED. Interpreting the estimated glomerular filtration rate in primary care: benefits and pitfalls. Cleve Clin J Med 2011; 78:189–195.
- National Kidney Foundation, Inc. KDOQI Clinical Practice Guidelines for Chronic Kidney Disease: Evaluation, Classification, and Stratification. http://www.kidney.org/professionals/kdoqi/guidelines_ckd/p4_class_g1.htm. Accessed March 5, 2014.
- Orlando LA, Owen WF, Matchar DB. Relationship between nephrologist care and progression of chronic kidney disease. N C Med J 2007; 68:9–16.
- Tseng CL, Kern EF, Miller DR, et al. Survival benefit of nephrologic care in patients with diabetes mellitus and chronic kidney disease. Arch Intern Med 2008; 168:55–62.
- Keith DS, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med 2004; 164:659–663.
- Serrano A, Huang J, Ghossein C, et al. Stabilization of glomerular filtration rate in advanced chronic kidney disease: a two-year follow-up of a cohort of chronic kidney disease patients stages 4 and 5. Adv Chronic Kidney Dis 2007; 14:105–112.
- Kinchen KS, Sadler J, Fink N, et al. The timing of specialist evaluation in chronic kidney disease and mortality. Ann Intern Med 2002; 137:479–486.
- Tonelli M, Muntner P, Lloyd A, et al; Alberta Kidney Disease Network. Risk of coronary events in people with chronic kidney disease compared with those with diabetes: a population-level cohort study. Lancet 2012; 380:807–814.
- Wattanakit K, Coresh J, Muntner P, Marsh J, Folsom AR. Cardiovascular risk among adults with chronic kidney disease, with or without prior myocardial infarction. J Am Coll Cardiol 2006; 48:1183–1189.
- Foley RN, Wang C, Collins AJ. Cardiovascular risk factor profiles and kidney function stage in the US general population: the NHANES III study. Mayo Clin Proc 2005; 80:1270–1277.
- Muntner P, He J, Astor BC, Folsom AR, Coresh J. Traditional and nontraditional risk factors predict coronary heart disease in chronic kidney disease: results from the Atherosclerosis Risk in Communities Study. J Am Soc Nephrol 2005; 16:529–538.
- Navaneethan SD, Schold JD, Kirwan JP, et al. Metabolic syndrome, ESRD, and death in CKD. Clin J Am Soc Nephrol 2013; 8:945–952.
- Foley RN, Murray AM, Li S, et al. Chronic kidney disease and the risk for cardiovascular disease, renal replacement, and death in the United States Medicare population, 1998 to 1999. J Am Soc Nephrol 2005; 16:489–495.
- Weiner DE, Tighiouart H, Amin MG, et al. Chronic kidney disease as a risk factor for cardiovascular disease and all-cause mortality: a pooled analysis of community-based studies. J Am Soc Nephrol 2004; 15:1307–1315.
- Gerstein HC, Mann JF, Yi Q, et al; HOPE Study Investigators. Albuminuria and risk of cardiovascular events, death, and heart failure in diabetic and nondiabetic individuals. JAMA 2001; 286:421–426.
- Hillege HL, Fidler V, Diercks GF, et al; Prevention of Renal and Vascular End Stage Disease (PREVEND) Study Group. Urinary albumin excretion predicts cardiovascular and noncardiovascular mortality in general population. Circulation 2002; 106:1777–1782.
- Foley RN, Collins AJ, Ishani A, Kalra PA. Calcium-phosphate levels and cardiovascular disease in community-dwelling adults: the Atherosclerosis Risk in Communities (ARIC) Study. Am Heart J 2008; 156:556–563.
- Tonelli M, Sacks F, Pfeffer M, Gao Z, Curhan G; Cholesterol And Recurrent Events Trial Investigators. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 2005; 112:2627–2633.
- Menon V, Wang X, Sarnak MJ, et al. Long-term outcomes in nondiabetic chronic kidney disease. Kidney Int 2008; 73:1310–1315.
- Kasiske BL. Hyperlipidemia in patients with chronic renal disease. Am J Kidney Dis 1998; 32(suppl 3):S142–S156.
- Kendrick J, Shlipak MG, Targher G, Cook T, Lindenfeld J, Chonchol M. Effect of lovastatin on primary prevention of cardiovascular events in mild CKD and kidney function loss: a post hoc analysis of the Air Force/Texas Coronary Atherosclerosis Prevention Study. Am J Kidney Dis 2010; 55:42–49.
- Colhoun HM, Betteridge DJ, Durrington PN, et al; CARDS Investigators. Effects of atorvastatin on kidney outcomes and cardiovascular disease in patients with diabetes: an analysis from the Collaborative Atorvastatin Diabetes Study (CARDS). Am J Kidney Dis 2009; 54:810–819.
- Koren MJ, Davidson MH, Wilson DJ, Fayyad RS, Zuckerman A, Reed DP; ALLIANCE Investigators. Focused atorvastatin therapy in managed-care patients with coronary heart disease and CKD. Am J Kidney Dis 2009; 53:741–750.
- Fellström BC, Jardine AG, Schmieder RE, et al; AURORA Study Group. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N Engl J Med 2009; 360:1395–1407.
- Chonchol M, Cook T, Kjekshus J, Pedersen TR, Lindenfeld J. Simvastatin for secondary prevention of all-cause mortality and major coronary events in patients with mild chronic renal insufficiency. Am J Kidney Dis 2007; 49:373–382.
- Ridker PM, MacFadyen J, Cressman M, Glynn RJ. Efficacy of rosuvastatin among men and women with moderate chronic kidney disease and elevated high-sensitivity C-reactive protein: a secondary analysis from the JUPITER (Justification for the Use of Statins in Prevention-an Intervention Trial Evaluating Rosuvastatin) trial. J Am Coll Cardiol 2010; 55:1266–1273.
- Baigent C, Landray MJ, Reith C, et al; SHARP Investigators. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet 2011; 377:2181–2192.
- Shepherd J, Kastelein JJ, Bittner V, et al; Treating to New Targets Investigators. Effect of intensive lipid lowering with atorvastatin on renal function in patients with coronary heart disease: the Treating to New Targets (TNT) study. Clin J Am Soc Nephrol 2007; 2:1131–1139.
- Tonelli M, Isles C, Craven T, et al. Effect of pravastatin on rate of kidney function loss in people with or at risk for coronary disease. Circulation 2005; 112:171–178.
- Palmer SC, Craig JC, Navaneethan SD, Tonelli M, Pellegrini F, Strippoli GF. Benefits and harms of statin therapy for persons with chronic kidney disease: a systematic review and meta-analysis. Ann Intern Med 2012; 157:263–275.
- National Kidney Foundation, Inc. KDOQI Clinical Practice Guidelines for Managing Dyslipidemias in Chronic Kidney Disease. http://www.kidney.org/professionals/kdoqi/guidelines_lipids/. Accessed March 5, 2014.
- Buckalew VM, Berg RL, Wang SR, Porush JG, Rauch S, Schulman G. Prevalence of hypertension in 1,795 subjects with chronic renal disease: the modification of diet in renal disease study baseline cohort. Modification of Diet in Renal Disease Study Group. Am J Kidney Dis 1996; 28:811–821.
- Coresh J, Wei GL, McQuillan G, et al. Prevalence of high blood pressure and elevated serum creatinine level in the United States: findings from the third National Health and Nutrition Examination Survey (1988–1994). Arch Intern Med 2001; 161:1207–1216.
- Klag MJ, Whelton PK, Randall BL, et al. Blood pressure and end-stage renal disease in men. N Engl J Med 1996; 334:13–18.
- Locatelli F, Marcelli D, Comelli M, et al. Proteinuria and blood pressure as causal components of progression to end-stage renal failure. Northern Italian Cooperative Study Group. Nephrol Dial Transplant 1996; 11:461–467.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- Jafar TH, Stark PC, Schmid CH, et al. Progression of chronic kidney disease: the role of blood pressure control, proteinuria, and angiotensin-converting enzyme inhibition: a patient-level meta-analysis. Ann Intern Med 2003; 139:244–252.
- Khosla N, Bakris G. Lessons learned from recent hypertension trials about kidney disease. Clin J Am Soc Nephrol 2006; 1:229–235.
- Norris KC, Greene T, Kopple J, et al. Baseline predictors of renal disease progression in the African American Study of Hypertension and Kidney Disease. J Am Soc Nephrol 2006; 17:2928–2936.
- Keane WF, Brenner BM, de Zeeuw D, et al; RENAAL Study Investigators. The risk of developing end-stage renal disease in patients with type 2 diabetes and nephropathy: the RENAAL study. Kidney Int 2003; 63:1499–1507.
- Ruggenenti P, Perna A, Mosconi L, et al. Proteinuria predicts end-stage renal failure in non-diabetic chronic nephropathies. The “Gruppo Italiano di Studi Epidemiologici in Nefrologia” (GISEN). Kidney Int Suppl 1997; 63:S54–S57.
- de Goeij MC, Liem M, de Jager DJ, et al; PREPARE-1 Study Group. Proteinuria as a risk marker for the progression of chronic kidney disease in patients on predialysis care and the role of angiotensin-converting enzyme inhibitor/angiotensin II receptor blocker treatment. Nephron Clin Pract 2012; 121:c73–c82.
- de Zeeuw D, Remuzzi G, Parving HH, et al. Albuminuria, a therapeutic target for cardiovascular protection in type 2 diabetic patients with nephropathy. Circulation 2004; 110:921–927.
- Ibsen H, Olsen MH, Wachtell K, et al. Reduction in albuminuria translates to reduction in cardiovascular events in hypertensive patients: losartan intervention for endpoint reduction in hypertension study. Hypertension 2005; 45:198–202.
- Atkins RC, Briganti EM, Lewis JB, et al. Proteinuria reduction and progression to renal failure in patients with type 2 diabetes mellitus and overt nephropathy. Am J Kidney Dis 2005; 45:281–287.
- Jafar TH, Stark PC, Schmid CH, et al; AIPRD Study Group; Angiotensin-Converting Enzyme Inhibition and Progression of Renal Disease. Proteinuria as a modifiable risk factor for the progression of non-diabetic renal disease. Kidney Int 2001; 60:1131–1140.
- ACE Inhibitors in Diabetic Nephropathy Trialist Group. Should all patients with type 1 diabetes mellitus and microalbuminuria receive angiotensin-converting enzyme inhibitors? A meta-analysis of individual patient data. Ann Intern Med 2001; 134:370–379.
- Casas JP, Chua W, Loukogeorgakis S, et al. Effect of inhibitors of the renin-angiotensin system and other antihypertensive drugs on renal outcomes: systematic review and meta-analysis. Lancet 2005; 366:2026–2033.
- Strippoli GF, Craig M, Deeks JJ, Schena FP, Craig JC. Effects of angiotensin converting enzyme inhibitors and angiotensin II receptor antagonists on mortality and renal outcomes in diabetic nephropathy: systematic review. BMJ 2004; 329:828.
- MacKinnon M, Shurraw S, Akbari A, Knoll GA, Jaffey J, Clark HD. Combination therapy with an angiotensin receptor blocker and an ACE inhibitor in proteinuric renal disease: a systematic review of the efficacy and safety data. Am J Kidney Dis 2006; 48:8–20.
- Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of mono-therapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med 2008; 148:30–48.
- Ruggenenti P, Perticucci E, Cravedi P, et al. Role of remission clinics in the longitudinal treatment of CKD. J Am Soc Nephrol 2008; 19:1213–1224.
- Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Esnault VL, Ekhlas A, Delcroix C, Moutel MG, Nguyen JM. Diuretic and enhanced sodium restriction results in improved antiproteinuric response to RAS blocking agents. J Am Soc Nephrol 2005; 16:474–481.
- Vogt L, Waanders F, Boomsma F, de Zeeuw D, Navis G. Effects of dietary sodium and hydrochlorothiazide on the antiproteinuric efficacy of losartan. J Am Soc Nephrol 2008; 19:999–1007.
- Ahmed AK, Kamath NS, El Kossi M, El Nahas AM. The impact of stopping inhibitors of the renin-angiotensin system in patients with advanced chronic kidney disease. Nephrol Dial Transplant 2010; 25:3977–3982.
- Bakris GL, Weir MR, Secic M, Campbell B, Weis-McNulty A. Differential effects of calcium antagonist subclasses on markers of nephropathy progression. Kidney Int 2004; 65:1991–2002.
- Kloke HJ, Wetzels JF, Koene RA, Huysmans FT. Effects of low-dose nifedipine on urinary protein excretion rate in patients with renal disease. Nephrol Dial Transplant 1998; 13:646–650.
- Shah SN, Abramowitz M, Hostetter TH, Melamed ML. Serum bicarbonate levels and the progression of kidney disease: a cohort study. Am J Kidney Dis 2009; 54:270–277.
- de Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol 2009; 20:2075–2084.
- Susantitaphong P, Sewaralthahab K, Balk EM, Jaber BL, Madias NE. Short- and long-term effects of alkali therapy in chronic kidney disease: a systematic review. Am J Nephrol 2012; 35:540–547.
- Nath KA, Hostetter MK, Hostetter TH. Ammonia-complement interaction in the pathogenesis of progressive renal injury. Kidney Int Suppl 1989; 27:S52–S54.
- Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am J Kidney Dis 2000; 35(suppl 2):S1–S140.
- Shimada T, Yamazaki Y, Takahashi M, et al. Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am J Physiol Renal Physiol 2005; 289:F1088–F1095.
- Hasegawa H, Nagano N, Urakawa I, et al. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int 2010; 78:975–980.
- de Boer IH, Rue TC, Kestenbaum B. Serum phosphorus concentrations in the third National Health and Nutrition Examination Survey (NHANES III). Am J Kidney Dis 2009; 53:399–407.
- Kendrick J, Cheung AK, Kaufman JS, et al; HOST Investigators. FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J Am Soc Nephrol 2011; 22:1913–1922.
- Palmer SC, Hayen A, Macaskill P, et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. JAMA. 2011; 305:1119–1127.
- Kooienga L, Fried L, Scragg R, Kendrick J, Smits G, Chonchol M. The effect of combined calcium and vitamin D3 supplementation on serum intact parathyroid hormone in moderate CKD. Am J Kidney Dis 2009; 53:408–416.
- National Kidney Foundation, Inc. KDOQI Clinical Practice Guidelines for Bone Metabolism and Disease in Chronic Kidney Disease. www.kidney.org/professionals/kdoqi/guidelines_bone/guide1.htm#table15. Accessed March 5, 2014.
- Kidney International. KDIGO Clinical Practice Guideline for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). http://kdigo.org/home/mineral-bone-disorder. Accessed March 5, 2014.
- Kazmi WH, Kausz AT, Khan S, et al. Anemia: an early complication of chronic renal insufficiency. Am J Kidney Dis 2001; 38:803–812.
- Sarnak MJ, Tighiouart H, Manjunath G, et al. Anemia as a risk factor for cardiovascular disease in the Atherosclerosis Risk in Communities (ARIC) study. J Am Coll Cardiol 2002; 40:27–33.
- Thorp ML, Johnson ES, Yang X, Petrik AF, Platt R, Smith DH. Effect of anaemia on mortality, cardiovascular hospitalizations and end-stage renal disease among patients with chronic kidney disease. Nephrology (Carlton) 2009; 14:240–246.
- Mircescu G, Gârneata L, Capusa C, Ursea N. Intravenous iron supplementation for the treatment of anaemia in pre-dialyzed chronic renal failure patients. Nephrol Dial Transplant 2006; 21:120–124.
- Silverberg DS, Iaina A, Peer G, et al. Intravenous iron supplementation for the treatment of the anemia of moderate to severe chronic renal failure patients not receiving dialysis. Am J Kidney Dis 1996; 27:234–238.
- Singh AK, Szczech L, Tang KL, et al; CHOIR Investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- Pfeffer MA, Burdmann EA, Chen CY, et al; TREAT Investigators. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 2009; 361:2019–2032.
- US Food and Drug Administration (FDA). FDA Drug Safety Communication: modified dosing recommendations to improve the safe use of erythropoiesis-stimulating agents (ESAs) in chronic kidney disease. http://www.fda.gov/drugs/drugsafety/ucm259639.htm. Accessed March 5, 2014.
- Kurella M, Covinsky KE, Collins AJ, Chertow GM. Octogenarians and nonagenarians starting dialysis in the United States. Ann Intern Med 2007; 146:177–183.
- Kurella Tamura M, Covinsky KE, Chertow GM, Yaffe K, Landefeld CS, McCulloch CE. Functional status of elderly adults before and after initiation of dialysis. N Engl J Med 2009; 361:1539–1547.
- Renal Physicians Association. Clinical Practice Guideline. Shared Decision-Making in the Appropriate Initiation of and Withdrawal from Dialysis. 2nd ed.
- Vollmer WM, Wahl PW, Blagg CR. Survival with dialysis and transplantation in patients with end-stage renal disease. N Engl J Med 1983; 308:1553–1558.
- Port FK, Wolfe RA, Mauger EA, Berling DP, Jiang K. Comparison of survival probabilities for dialysis patients vs cadaveric renal transplant recipients. JAMA 1993; 270:1339–1343.
- Wolfe RA, Ashby VB, Milford EL, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med 1999; 341:1725–1730.
- Cosio FG, Alamir A, Yim S, et al. Patient survival after renal transplantation: I. The impact of dialysis pre-transplant. Kidney Int 1998; 53:767–772.
- Meier-Kriesche HU, Port FK, Ojo AO, et al. Effect of waiting time on renal transplant outcome. Kidney Int 2000; 58:1311–1317.
- Mange KC, Joffe MM, Feldman HI. Effect of the use or nonuse of long-term dialysis on the subsequent survival of renal transplants from living donors. N Engl J Med 2001; 344:726–731.
- Dhingra RK, Young EW, Hulbert-Shearon TE, Leavey SF, Port FK. Type of vascular access and mortality in US hemodialysis patients. Kidney Int 2001; 60:1443–1451.
- Nassar GM, Ayus JC. Infectious complications of the hemodialysis access. Kidney Int 2001; 60:1–13.
- Perera GB, Mueller MP, Kubaska SM, Wilson SE, Lawrence PF, Fujitani RM. Superiority of autogenous arteriovenous hemodialysis access: maintenance of function with fewer secondary interventions. Ann Vasc Surg 2004; 18:66–73.
- Basile C, Casucci F, Lomonte C. Timing of first cannulation of arteriovenous fistula: time matters, but there is also something else. Nephrol Dial Transplant 2005; 20:1519–1520.
- Biuckians A, Scott EC, Meier GH, Panneton JM, Glickman MH. The natural history of autologous fistulas as first-time dialysis access in the KDOQI era. J Vasc Surg 2008; 47:415–421.
- National Kidney Foundation, Inc. KDOQI Clinical Practice Guidelines for Vascular Access. http://www.kidney.org/professionals/KDOQI/guideline_upHD_PD_VA/va_guide1.htm. Accessed March 5, 2014.
- Arora P, Obrador GT, Ruthazer R, et al. Prevalence, predictors, and consequences of late nephrology referral at a tertiary care center. J Am Soc Nephrol 1999; 10:1281–1286.
- Gøransson LG, Bergrem H. Consequences of late referral of patients with end-stage renal disease. J Intern Med 2001; 250:154–159.
- Cooper BA, Branley P, Bulfone L, et al. A randomized, controlled trial of early versus late initiation of dialysis. N Engl J Med 2010; 363:609–619.
- Carson RC, Juszczak M, Davenport A, Burns A. Is maximum conservative management an equivalent treatment option to dialysis for elderly patients with significant comorbid disease? Clin J Am Soc Nephrol 2009; 4:1611–1619.
- Murtagh FE, Marsh JE, Donohoe P, Ekbal NJ, Sheerin NS, Harris FE. Dialysis or not? A comparative survival study of patients over 75 years with chronic kidney disease stage 5. Nephrol Dial Transplant 2007; 22:1955–1962.
- United States Renal Data System (USRDS). Identification and care of patients with CKD. http://www.usrds.org/2012/pdf/v1_ch2_12.pdf. Accessed March 5, 2014.
- Simon J, Amde M, Poggio ED. Interpreting the estimated glomerular filtration rate in primary care: benefits and pitfalls. Cleve Clin J Med 2011; 78:189–195.
- National Kidney Foundation, Inc. KDOQI Clinical Practice Guidelines for Chronic Kidney Disease: Evaluation, Classification, and Stratification. http://www.kidney.org/professionals/kdoqi/guidelines_ckd/p4_class_g1.htm. Accessed March 5, 2014.
- Orlando LA, Owen WF, Matchar DB. Relationship between nephrologist care and progression of chronic kidney disease. N C Med J 2007; 68:9–16.
- Tseng CL, Kern EF, Miller DR, et al. Survival benefit of nephrologic care in patients with diabetes mellitus and chronic kidney disease. Arch Intern Med 2008; 168:55–62.
- Keith DS, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med 2004; 164:659–663.
- Serrano A, Huang J, Ghossein C, et al. Stabilization of glomerular filtration rate in advanced chronic kidney disease: a two-year follow-up of a cohort of chronic kidney disease patients stages 4 and 5. Adv Chronic Kidney Dis 2007; 14:105–112.
- Kinchen KS, Sadler J, Fink N, et al. The timing of specialist evaluation in chronic kidney disease and mortality. Ann Intern Med 2002; 137:479–486.
- Tonelli M, Muntner P, Lloyd A, et al; Alberta Kidney Disease Network. Risk of coronary events in people with chronic kidney disease compared with those with diabetes: a population-level cohort study. Lancet 2012; 380:807–814.
- Wattanakit K, Coresh J, Muntner P, Marsh J, Folsom AR. Cardiovascular risk among adults with chronic kidney disease, with or without prior myocardial infarction. J Am Coll Cardiol 2006; 48:1183–1189.
- Foley RN, Wang C, Collins AJ. Cardiovascular risk factor profiles and kidney function stage in the US general population: the NHANES III study. Mayo Clin Proc 2005; 80:1270–1277.
- Muntner P, He J, Astor BC, Folsom AR, Coresh J. Traditional and nontraditional risk factors predict coronary heart disease in chronic kidney disease: results from the Atherosclerosis Risk in Communities Study. J Am Soc Nephrol 2005; 16:529–538.
- Navaneethan SD, Schold JD, Kirwan JP, et al. Metabolic syndrome, ESRD, and death in CKD. Clin J Am Soc Nephrol 2013; 8:945–952.
- Foley RN, Murray AM, Li S, et al. Chronic kidney disease and the risk for cardiovascular disease, renal replacement, and death in the United States Medicare population, 1998 to 1999. J Am Soc Nephrol 2005; 16:489–495.
- Weiner DE, Tighiouart H, Amin MG, et al. Chronic kidney disease as a risk factor for cardiovascular disease and all-cause mortality: a pooled analysis of community-based studies. J Am Soc Nephrol 2004; 15:1307–1315.
- Gerstein HC, Mann JF, Yi Q, et al; HOPE Study Investigators. Albuminuria and risk of cardiovascular events, death, and heart failure in diabetic and nondiabetic individuals. JAMA 2001; 286:421–426.
- Hillege HL, Fidler V, Diercks GF, et al; Prevention of Renal and Vascular End Stage Disease (PREVEND) Study Group. Urinary albumin excretion predicts cardiovascular and noncardiovascular mortality in general population. Circulation 2002; 106:1777–1782.
- Foley RN, Collins AJ, Ishani A, Kalra PA. Calcium-phosphate levels and cardiovascular disease in community-dwelling adults: the Atherosclerosis Risk in Communities (ARIC) Study. Am Heart J 2008; 156:556–563.
- Tonelli M, Sacks F, Pfeffer M, Gao Z, Curhan G; Cholesterol And Recurrent Events Trial Investigators. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 2005; 112:2627–2633.
- Menon V, Wang X, Sarnak MJ, et al. Long-term outcomes in nondiabetic chronic kidney disease. Kidney Int 2008; 73:1310–1315.
- Kasiske BL. Hyperlipidemia in patients with chronic renal disease. Am J Kidney Dis 1998; 32(suppl 3):S142–S156.
- Kendrick J, Shlipak MG, Targher G, Cook T, Lindenfeld J, Chonchol M. Effect of lovastatin on primary prevention of cardiovascular events in mild CKD and kidney function loss: a post hoc analysis of the Air Force/Texas Coronary Atherosclerosis Prevention Study. Am J Kidney Dis 2010; 55:42–49.
- Colhoun HM, Betteridge DJ, Durrington PN, et al; CARDS Investigators. Effects of atorvastatin on kidney outcomes and cardiovascular disease in patients with diabetes: an analysis from the Collaborative Atorvastatin Diabetes Study (CARDS). Am J Kidney Dis 2009; 54:810–819.
- Koren MJ, Davidson MH, Wilson DJ, Fayyad RS, Zuckerman A, Reed DP; ALLIANCE Investigators. Focused atorvastatin therapy in managed-care patients with coronary heart disease and CKD. Am J Kidney Dis 2009; 53:741–750.
- Fellström BC, Jardine AG, Schmieder RE, et al; AURORA Study Group. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N Engl J Med 2009; 360:1395–1407.
- Chonchol M, Cook T, Kjekshus J, Pedersen TR, Lindenfeld J. Simvastatin for secondary prevention of all-cause mortality and major coronary events in patients with mild chronic renal insufficiency. Am J Kidney Dis 2007; 49:373–382.
- Ridker PM, MacFadyen J, Cressman M, Glynn RJ. Efficacy of rosuvastatin among men and women with moderate chronic kidney disease and elevated high-sensitivity C-reactive protein: a secondary analysis from the JUPITER (Justification for the Use of Statins in Prevention-an Intervention Trial Evaluating Rosuvastatin) trial. J Am Coll Cardiol 2010; 55:1266–1273.
- Baigent C, Landray MJ, Reith C, et al; SHARP Investigators. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet 2011; 377:2181–2192.
- Shepherd J, Kastelein JJ, Bittner V, et al; Treating to New Targets Investigators. Effect of intensive lipid lowering with atorvastatin on renal function in patients with coronary heart disease: the Treating to New Targets (TNT) study. Clin J Am Soc Nephrol 2007; 2:1131–1139.
- Tonelli M, Isles C, Craven T, et al. Effect of pravastatin on rate of kidney function loss in people with or at risk for coronary disease. Circulation 2005; 112:171–178.
- Palmer SC, Craig JC, Navaneethan SD, Tonelli M, Pellegrini F, Strippoli GF. Benefits and harms of statin therapy for persons with chronic kidney disease: a systematic review and meta-analysis. Ann Intern Med 2012; 157:263–275.
- National Kidney Foundation, Inc. KDOQI Clinical Practice Guidelines for Managing Dyslipidemias in Chronic Kidney Disease. http://www.kidney.org/professionals/kdoqi/guidelines_lipids/. Accessed March 5, 2014.
- Buckalew VM, Berg RL, Wang SR, Porush JG, Rauch S, Schulman G. Prevalence of hypertension in 1,795 subjects with chronic renal disease: the modification of diet in renal disease study baseline cohort. Modification of Diet in Renal Disease Study Group. Am J Kidney Dis 1996; 28:811–821.
- Coresh J, Wei GL, McQuillan G, et al. Prevalence of high blood pressure and elevated serum creatinine level in the United States: findings from the third National Health and Nutrition Examination Survey (1988–1994). Arch Intern Med 2001; 161:1207–1216.
- Klag MJ, Whelton PK, Randall BL, et al. Blood pressure and end-stage renal disease in men. N Engl J Med 1996; 334:13–18.
- Locatelli F, Marcelli D, Comelli M, et al. Proteinuria and blood pressure as causal components of progression to end-stage renal failure. Northern Italian Cooperative Study Group. Nephrol Dial Transplant 1996; 11:461–467.
- ACCORD Study Group; Cushman WC, Evans GW, Byington RP, et al. Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- Jafar TH, Stark PC, Schmid CH, et al. Progression of chronic kidney disease: the role of blood pressure control, proteinuria, and angiotensin-converting enzyme inhibition: a patient-level meta-analysis. Ann Intern Med 2003; 139:244–252.
- Khosla N, Bakris G. Lessons learned from recent hypertension trials about kidney disease. Clin J Am Soc Nephrol 2006; 1:229–235.
- Norris KC, Greene T, Kopple J, et al. Baseline predictors of renal disease progression in the African American Study of Hypertension and Kidney Disease. J Am Soc Nephrol 2006; 17:2928–2936.
- Keane WF, Brenner BM, de Zeeuw D, et al; RENAAL Study Investigators. The risk of developing end-stage renal disease in patients with type 2 diabetes and nephropathy: the RENAAL study. Kidney Int 2003; 63:1499–1507.
- Ruggenenti P, Perna A, Mosconi L, et al. Proteinuria predicts end-stage renal failure in non-diabetic chronic nephropathies. The “Gruppo Italiano di Studi Epidemiologici in Nefrologia” (GISEN). Kidney Int Suppl 1997; 63:S54–S57.
- de Goeij MC, Liem M, de Jager DJ, et al; PREPARE-1 Study Group. Proteinuria as a risk marker for the progression of chronic kidney disease in patients on predialysis care and the role of angiotensin-converting enzyme inhibitor/angiotensin II receptor blocker treatment. Nephron Clin Pract 2012; 121:c73–c82.
- de Zeeuw D, Remuzzi G, Parving HH, et al. Albuminuria, a therapeutic target for cardiovascular protection in type 2 diabetic patients with nephropathy. Circulation 2004; 110:921–927.
- Ibsen H, Olsen MH, Wachtell K, et al. Reduction in albuminuria translates to reduction in cardiovascular events in hypertensive patients: losartan intervention for endpoint reduction in hypertension study. Hypertension 2005; 45:198–202.
- Atkins RC, Briganti EM, Lewis JB, et al. Proteinuria reduction and progression to renal failure in patients with type 2 diabetes mellitus and overt nephropathy. Am J Kidney Dis 2005; 45:281–287.
- Jafar TH, Stark PC, Schmid CH, et al; AIPRD Study Group; Angiotensin-Converting Enzyme Inhibition and Progression of Renal Disease. Proteinuria as a modifiable risk factor for the progression of non-diabetic renal disease. Kidney Int 2001; 60:1131–1140.
- ACE Inhibitors in Diabetic Nephropathy Trialist Group. Should all patients with type 1 diabetes mellitus and microalbuminuria receive angiotensin-converting enzyme inhibitors? A meta-analysis of individual patient data. Ann Intern Med 2001; 134:370–379.
- Casas JP, Chua W, Loukogeorgakis S, et al. Effect of inhibitors of the renin-angiotensin system and other antihypertensive drugs on renal outcomes: systematic review and meta-analysis. Lancet 2005; 366:2026–2033.
- Strippoli GF, Craig M, Deeks JJ, Schena FP, Craig JC. Effects of angiotensin converting enzyme inhibitors and angiotensin II receptor antagonists on mortality and renal outcomes in diabetic nephropathy: systematic review. BMJ 2004; 329:828.
- MacKinnon M, Shurraw S, Akbari A, Knoll GA, Jaffey J, Clark HD. Combination therapy with an angiotensin receptor blocker and an ACE inhibitor in proteinuric renal disease: a systematic review of the efficacy and safety data. Am J Kidney Dis 2006; 48:8–20.
- Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of mono-therapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med 2008; 148:30–48.
- Ruggenenti P, Perticucci E, Cravedi P, et al. Role of remission clinics in the longitudinal treatment of CKD. J Am Soc Nephrol 2008; 19:1213–1224.
- Mann JF, Schmieder RE, McQueen M, et al; ONTARGET investigators. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372:547–553.
- Esnault VL, Ekhlas A, Delcroix C, Moutel MG, Nguyen JM. Diuretic and enhanced sodium restriction results in improved antiproteinuric response to RAS blocking agents. J Am Soc Nephrol 2005; 16:474–481.
- Vogt L, Waanders F, Boomsma F, de Zeeuw D, Navis G. Effects of dietary sodium and hydrochlorothiazide on the antiproteinuric efficacy of losartan. J Am Soc Nephrol 2008; 19:999–1007.
- Ahmed AK, Kamath NS, El Kossi M, El Nahas AM. The impact of stopping inhibitors of the renin-angiotensin system in patients with advanced chronic kidney disease. Nephrol Dial Transplant 2010; 25:3977–3982.
- Bakris GL, Weir MR, Secic M, Campbell B, Weis-McNulty A. Differential effects of calcium antagonist subclasses on markers of nephropathy progression. Kidney Int 2004; 65:1991–2002.
- Kloke HJ, Wetzels JF, Koene RA, Huysmans FT. Effects of low-dose nifedipine on urinary protein excretion rate in patients with renal disease. Nephrol Dial Transplant 1998; 13:646–650.
- Shah SN, Abramowitz M, Hostetter TH, Melamed ML. Serum bicarbonate levels and the progression of kidney disease: a cohort study. Am J Kidney Dis 2009; 54:270–277.
- de Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol 2009; 20:2075–2084.
- Susantitaphong P, Sewaralthahab K, Balk EM, Jaber BL, Madias NE. Short- and long-term effects of alkali therapy in chronic kidney disease: a systematic review. Am J Nephrol 2012; 35:540–547.
- Nath KA, Hostetter MK, Hostetter TH. Ammonia-complement interaction in the pathogenesis of progressive renal injury. Kidney Int Suppl 1989; 27:S52–S54.
- Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am J Kidney Dis 2000; 35(suppl 2):S1–S140.
- Shimada T, Yamazaki Y, Takahashi M, et al. Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am J Physiol Renal Physiol 2005; 289:F1088–F1095.
- Hasegawa H, Nagano N, Urakawa I, et al. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int 2010; 78:975–980.
- de Boer IH, Rue TC, Kestenbaum B. Serum phosphorus concentrations in the third National Health and Nutrition Examination Survey (NHANES III). Am J Kidney Dis 2009; 53:399–407.
- Kendrick J, Cheung AK, Kaufman JS, et al; HOST Investigators. FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J Am Soc Nephrol 2011; 22:1913–1922.
- Palmer SC, Hayen A, Macaskill P, et al. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: a systematic review and meta-analysis. JAMA. 2011; 305:1119–1127.
- Kooienga L, Fried L, Scragg R, Kendrick J, Smits G, Chonchol M. The effect of combined calcium and vitamin D3 supplementation on serum intact parathyroid hormone in moderate CKD. Am J Kidney Dis 2009; 53:408–416.
- National Kidney Foundation, Inc. KDOQI Clinical Practice Guidelines for Bone Metabolism and Disease in Chronic Kidney Disease. www.kidney.org/professionals/kdoqi/guidelines_bone/guide1.htm#table15. Accessed March 5, 2014.
- Kidney International. KDIGO Clinical Practice Guideline for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). http://kdigo.org/home/mineral-bone-disorder. Accessed March 5, 2014.
- Kazmi WH, Kausz AT, Khan S, et al. Anemia: an early complication of chronic renal insufficiency. Am J Kidney Dis 2001; 38:803–812.
- Sarnak MJ, Tighiouart H, Manjunath G, et al. Anemia as a risk factor for cardiovascular disease in the Atherosclerosis Risk in Communities (ARIC) study. J Am Coll Cardiol 2002; 40:27–33.
- Thorp ML, Johnson ES, Yang X, Petrik AF, Platt R, Smith DH. Effect of anaemia on mortality, cardiovascular hospitalizations and end-stage renal disease among patients with chronic kidney disease. Nephrology (Carlton) 2009; 14:240–246.
- Mircescu G, Gârneata L, Capusa C, Ursea N. Intravenous iron supplementation for the treatment of anaemia in pre-dialyzed chronic renal failure patients. Nephrol Dial Transplant 2006; 21:120–124.
- Silverberg DS, Iaina A, Peer G, et al. Intravenous iron supplementation for the treatment of the anemia of moderate to severe chronic renal failure patients not receiving dialysis. Am J Kidney Dis 1996; 27:234–238.
- Singh AK, Szczech L, Tang KL, et al; CHOIR Investigators. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006; 355:2085–2098.
- Drüeke TB, Locatelli F, Clyne N, et al; CREATE Investigators. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med 2006; 355:2071–2084.
- Pfeffer MA, Burdmann EA, Chen CY, et al; TREAT Investigators. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med 2009; 361:2019–2032.
- US Food and Drug Administration (FDA). FDA Drug Safety Communication: modified dosing recommendations to improve the safe use of erythropoiesis-stimulating agents (ESAs) in chronic kidney disease. http://www.fda.gov/drugs/drugsafety/ucm259639.htm. Accessed March 5, 2014.
- Kurella M, Covinsky KE, Collins AJ, Chertow GM. Octogenarians and nonagenarians starting dialysis in the United States. Ann Intern Med 2007; 146:177–183.
- Kurella Tamura M, Covinsky KE, Chertow GM, Yaffe K, Landefeld CS, McCulloch CE. Functional status of elderly adults before and after initiation of dialysis. N Engl J Med 2009; 361:1539–1547.
- Renal Physicians Association. Clinical Practice Guideline. Shared Decision-Making in the Appropriate Initiation of and Withdrawal from Dialysis. 2nd ed.
- Vollmer WM, Wahl PW, Blagg CR. Survival with dialysis and transplantation in patients with end-stage renal disease. N Engl J Med 1983; 308:1553–1558.
- Port FK, Wolfe RA, Mauger EA, Berling DP, Jiang K. Comparison of survival probabilities for dialysis patients vs cadaveric renal transplant recipients. JAMA 1993; 270:1339–1343.
- Wolfe RA, Ashby VB, Milford EL, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med 1999; 341:1725–1730.
- Cosio FG, Alamir A, Yim S, et al. Patient survival after renal transplantation: I. The impact of dialysis pre-transplant. Kidney Int 1998; 53:767–772.
- Meier-Kriesche HU, Port FK, Ojo AO, et al. Effect of waiting time on renal transplant outcome. Kidney Int 2000; 58:1311–1317.
- Mange KC, Joffe MM, Feldman HI. Effect of the use or nonuse of long-term dialysis on the subsequent survival of renal transplants from living donors. N Engl J Med 2001; 344:726–731.
- Dhingra RK, Young EW, Hulbert-Shearon TE, Leavey SF, Port FK. Type of vascular access and mortality in US hemodialysis patients. Kidney Int 2001; 60:1443–1451.
- Nassar GM, Ayus JC. Infectious complications of the hemodialysis access. Kidney Int 2001; 60:1–13.
- Perera GB, Mueller MP, Kubaska SM, Wilson SE, Lawrence PF, Fujitani RM. Superiority of autogenous arteriovenous hemodialysis access: maintenance of function with fewer secondary interventions. Ann Vasc Surg 2004; 18:66–73.
- Basile C, Casucci F, Lomonte C. Timing of first cannulation of arteriovenous fistula: time matters, but there is also something else. Nephrol Dial Transplant 2005; 20:1519–1520.
- Biuckians A, Scott EC, Meier GH, Panneton JM, Glickman MH. The natural history of autologous fistulas as first-time dialysis access in the KDOQI era. J Vasc Surg 2008; 47:415–421.
- National Kidney Foundation, Inc. KDOQI Clinical Practice Guidelines for Vascular Access. http://www.kidney.org/professionals/KDOQI/guideline_upHD_PD_VA/va_guide1.htm. Accessed March 5, 2014.
- Arora P, Obrador GT, Ruthazer R, et al. Prevalence, predictors, and consequences of late nephrology referral at a tertiary care center. J Am Soc Nephrol 1999; 10:1281–1286.
- Gøransson LG, Bergrem H. Consequences of late referral of patients with end-stage renal disease. J Intern Med 2001; 250:154–159.
- Cooper BA, Branley P, Bulfone L, et al. A randomized, controlled trial of early versus late initiation of dialysis. N Engl J Med 2010; 363:609–619.
- Carson RC, Juszczak M, Davenport A, Burns A. Is maximum conservative management an equivalent treatment option to dialysis for elderly patients with significant comorbid disease? Clin J Am Soc Nephrol 2009; 4:1611–1619.
- Murtagh FE, Marsh JE, Donohoe P, Ekbal NJ, Sheerin NS, Harris FE. Dialysis or not? A comparative survival study of patients over 75 years with chronic kidney disease stage 5. Nephrol Dial Transplant 2007; 22:1955–1962.
KEY POINTS
- Steps to stabilize renal function include blood pressure and diabetes control.
- Patients have a very high risk of cardiovascular disease, and one should try to reduce modifiable risk factors such as hypertension (which is also a risk factor for the progression of CKD) and hyperlipidemia.
- In addition to controlling blood pressure, angiotensin-converting enzyme inhibitors and angiotensin receptor blockers reduce proteinuria, a risk factor for progression of CKD.
- Patients with CKD develop secondary hyperparathyroidism, hyperphosphatemia, and, in advanced CKD, hypocalcemia, all leading to disorders of bone mineral metabolism. Low vitamin D levels should be raised with supplements, and high phosphorus levels should be lowered with dietary restriction and phosphate binders.
Stenting atherosclerotic renal arteries: Time to be less aggressive
Author’s note: Atherosclerosis accounts for about 90% of cases of renal artery stenosis in people over age 40.1 Fibromuscular dysplasia, the other major cause, is a separate topic; in this paper “renal artery stenosis” refers to atherosclerotic disease only.
Renal artery stenosis is very common, and the number of angioplasty-stenting procedures performed every year is on the rise. Yet there is no overwhelming evidence that intervention yields clinical benefits—ie, better blood pressure control or renal function— than does medical therapy.
Earlier randomized controlled trials comparing angioplasty without stents and medical management showed no impressive difference in blood pressure.2,3 The data on renal function were even more questionable, with some studies suggesting that, with stenting, the chance of worsening renal function is equal to that of improvement.4
Two large randomized trials comparing renal intervention with medical therapy failed to show any benefit of intervention.5–7 A third study is under way.8
It is time to strongly reconsider the current aggressive approach to revascularization of stenotic renal arteries and take a more coordinated, critical approach.
RENAL INTERVENTIONS ON THE RISE
Renal angioplasty began replacing surgical revascularization in the 1990s, as this less-invasive procedure became more readily available and was shown to have similar clinical outcomes.9 In the last decade, stent placement during angioplasty has become standard, improving the rates of technical success.
As these procedures have become easier to perform and their radiographic outcomes have become more consistent, interventionalists have become more likely, if they see stenosis in a renal artery, to intervene and insert a stent, regardless of proven benefit. In addition, interventionalists from at least three different specialties now compete for these procedures, often by looking at the renal arteries during angiography of other vascular beds (the “drive-by”).
As a result, the number of renal interventions has been rising. Medicare received 21,660 claims for renal artery interventions (surgery or angioplasty) in 2000, compared with 13,380 in 1996—an increase of 62%. However, the number of surgeries actually decreased by 45% during this time, while the number of percutaneous procedures increased by 240%. The number of endovascular claims for renal artery stenosis by cardiologists alone rose 390%.10 Since then, the reports on intervention have been mixed, with one report citing a continued increase in 2005 to 35,000 claims,11 and another suggesting a decrease back to 1997 levels.12
HOW COMMON IS RENAL ARTERY STENOSIS?
The prevalence of renal artery stenosis depends on the definition used and the population screened. It is more common in older patients who have risk factors for other vascular diseases than in the general population.
Renal Doppler ultrasonography can detect stenosis only if the artery is narrowed by more than 60%. Hansen et al13 used ultrasonography to screen 870 people over age 65 and found a lesion (a narrowing of more than 60%) in 6.8%.
Angiography (direct, computed tomographic, or magnetic resonance) can detect less-severe stenosis. Thus, most angiographic studies define renal artery stenosis as a narrowing of more than 50%, and severe disease as a narrowing of more than 70%. Many experts believe that unilateral stenosis needs to be more than 70% to pose a risk to the kidney.14,15
Angiographic prevalence studies have been performed only in patients who were undergoing angiography for another reason such as coronary or peripheral arterial disease that inherently places them at higher risk of renal artery stenosis. For instance, renal artery stenosis is found in 11% to 28% of patients undergoing diagnostic cardiac catheterization. 16
No studies of the prevalence of renal artery stenosis have been performed in the general population. Medicare data indicate that from 1999 to 2001 the incidence of diagnosed renal artery stenosis was 3.7 per 1,000 patientyears. 17 Holley et al,18 in an autopsy series, found renal artery stenosis of greater than 50% in 27% of patients over age 50 and in 56.4% of hypertensive patients. The prevalence was 10% in normotensive patients.
WHO IS AT RISK?
Factors associated with a higher risk of finding renal artery stenosis on a radiographic study include14:
- Older age
- Female sex
- Hypertension
- Three-vessel coronary artery disease
- Peripheral artery disease
- Chronic kidney disease
- Diabetes

- A low level of high-density lipoprotein cholesterol
- The use of at least two cardiovascular drugs.
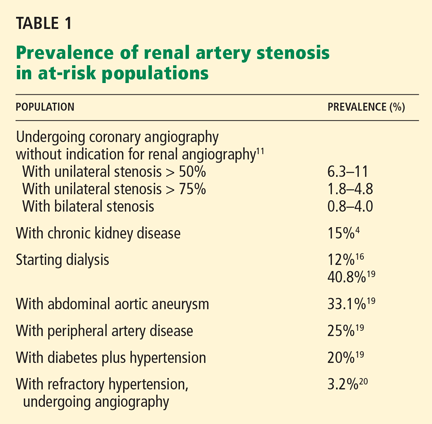
The prevalence of renal artery stenosis in at-risk populations ranges from 3% to 75% (Table 1).2,4,6,19,20
HOW OFTEN DOES STENOSIS PROGRESS?
The reported rates of progression of atherosclerotic renal artery lesions vary depending on the type of imaging test used and the reason for doing it.
In studies that used duplex ultrasonography, roughly half of lesions smaller than 60% grew to greater than 60% over 3 years.21,22 The risk of total occlusion of an artery was relatively low and depended on the severity of stenosis: 0.7% if the baseline stenosis was less than 60% and 2.3% to 7% if it was greater.21,22
In a seminal study in 1984, Schreiber and colleagues23 compared serial angiograms obtained a mean of 52 months apart in 85 patients who did not undergo intervention. Stenosis had progressed in 37 (44%), and to the point of total occlusion in 14 (16%). In contrast, a 1998 study found progression in 11.1% over 2.6 years, with older patients, women, and those with baseline coronary artery disease at higher risk.24
The the rates of progression differed in these two studies probably because the indications for screening were different (clinical suspicion23 vs routine screening during coronary angiography24), as was the severity of stenosis at the time of diagnosis. Also, when these studies were done, fewer people were taking statins. Thus, similar studies, if repeated, might show even lower rates of progression.
Finally, progression of renal artery stenosis has not been correlated with worsening renal function.
FOUR CLINICAL PRESENTATIONS OF RENAL ARTERY STENOSIS
Renal artery stenosis can present in one of four ways:
Clinically silent stenosis. Because renal artery stenosis is most often found in older patients, who are more likely to have essential hypertension and chronic kidney disease due to other causes, it can be an incidental finding that is completely clinically silent.16,25
Renovascular hypertension is defined as high blood pressure due to up-regulation of neurohormonal activity in response to decreased perfusion from renal artery stenosis. Renal artery stenosis is estimated to be the cause of hypertension in only 0.5% to 4.0% of hypertensive patients, or in 26% of patients with secondary hypertension.3
Ischemic nephropathy is more difficult to define because ischemia alone rarely explains the damage done to the kidneys. Activation of neurohormonal pathways and microvascular injury are thought to contribute to oxidative stress and fibrosis.26 These phenomena may explain why similar degrees of stenosis lead to varying degrees of kidney damage in different patients and why the severity of stenosis does not correlate with the degree of renal dysfunction.27
Furthermore, stenosis may lead to irreversible but stable kidney damage. It is therefore not surprising that, in studies in unselected populations (ie, studies that included patients with all presentations of renal artery stenosis, not just those more likely to benefit), up to two-thirds of renal interventions yielded no clinical benefit.25
As a result, if we define ischemic nephropathy as renal artery stenosis with renal dysfunction not attributable to another cause, we probably will overestimate the prevalence of ischemic nephropathy, leading to overly optimistic expectations about the response to revascularization.
Recurrent “flash” pulmonary edema is a less common presentation, usually occurring in patients with critical bilateral renal artery stenosis or unilateral stenosis in an artery supplying a solitary functioning kidney. Most have severe hypertension (average systolic blood pressure 174–207 mm Hg) and poor renal function.28–30
The association between pulmonary edema and bilateral renal artery stenosis was first noted in 1998 by Pickering et al,31 who in several case series showed that 82% to 92% of patients with recurrent pulmonary edema and renal artery stenosis had bilateral stenosis, compared with 20% to 65% of those with other presentations. Later case series corroborated this finding: 85% to 100% of patients with renal artery stenosis and pulmonary edema had bilateral stenosis.28–30
STENTING IS NOW STANDARD
Stenting has become standard in the endovascular treatment of renal artery stenosis.
Most atherosclerotic renal artery lesions are located in the ostium (ie, where the artery branches off from the aorta), and many are extensions of calcified aortic plaque.26,32 These hard lesions tend to rebound to their original shape more often with balloon angioplasty alone. Stenting provides the additional force needed to permanently disrupt the lesion, leading to a longer-lasting result.
Rates of technical success (dilating the artery during the intervention) are higher with stents than without them (98% vs 46%– 77%).33,34 If the lesion is ostial, this difference is even more impressive (75% vs 29%). In addition, restenosis rates at 6 months are lower with stents (14% vs 26%–48%).34
GOALS: LOWER THE BLOOD PRESSURE, SAVE THE KIDNEY
Because endovascular procedures pose some risk to the patient, it is critical to intervene only in patients most likely to respond clinically. The decision to intervene depends largely on the clinical goal, which should depend on the clinical presentation.

However, if renal artery stenosis is clinically silent, most of the evidence suggests that intervention has no benefit. Furthermore, although retrospective studies have indicated that intervention may improve survival rates,35,36 prospective studies have not. Similarly, studies have not shown that intervention generally improves cardiovascular outcomes, even though renal artery stenosis is associated with cardiovascular risk.
Hypertension plus stenosis is not necessarily renovascular hypertension
Essential hypertension and clinically silent renal artery stenosis often coexist, which is why blood pressure control often does not improve after stenting. Also, essential hypertension often coexists with renovascular hypertension.37 In this situation, stenting may not eliminate the need for antihypertensive drugs, although it may improve blood pressure control and decrease the drug burden.
Before stents came into use, several randomized controlled trials found that blood pressure was no better controlled after angioplasty, 2,3,38 except in cases of bilateral stenosis.2 This may be because stenosis tended to recur after angioplasty without stents.
The 2000 Dutch Renal Artery Stenosis Intervention Cooperative (DRASTIC) study was the first randomized controlled trial to examine the effect of angioplasty on blood pressure control in renal artery stenosis.38 It had significant design flaws. For example, many patients crossed over from the medical management group to the intervention group because their hypertension was resistant to medical therapy. Overall, intervention (balloon angioplasty without stents in 54 of 56 patients, with stents in the other 2) carried no benefit. However, in subgroup analysis, the patients who crossed over because of resistant hypertension (failure of a three-drug regimen) were more likely to benefit from angioplasty. This suggested that risk stratification should take place early on, before proceeding with revascularization.
With stents, Zeller,39 in a prospective nonrandomized study, found that the mean arterial pressure decreased by 10 mm Hg. Randomized trials (see below) have failed to demonstrate such a benefit.
Stenting may not improve renal function
Coincidental renal artery stenosis in a patient with unrelated chronic kidney disease is very hard to differentiate from true ischemic nephropathy. Furthermore, most patients with ischemic nephropathy do not benefit from revascularization, making it challenging to identify those few whose renal function may respond.
Given that patients with chronic kidney disease tend to have a higher risk of cardiovascular disease, it is not surprising that 15% of them may have renal artery stenosis,4 most often incidental.
Chábová40 examined the outcomes of 68 patients who had chronic kidney disease and a renal artery lesion larger than 70% who did not undergo angioplasty. In only 10 (15%) of the patients did the glomerular filtration rate (GFR) decline by more than 50% of its baseline value during the study period of 3 years. Given the retrospective nature of the study, it cannot be determined (and is rather unlikely) that ischemic nephropathy was the cause of the decline in kidney function in all 10 patients.
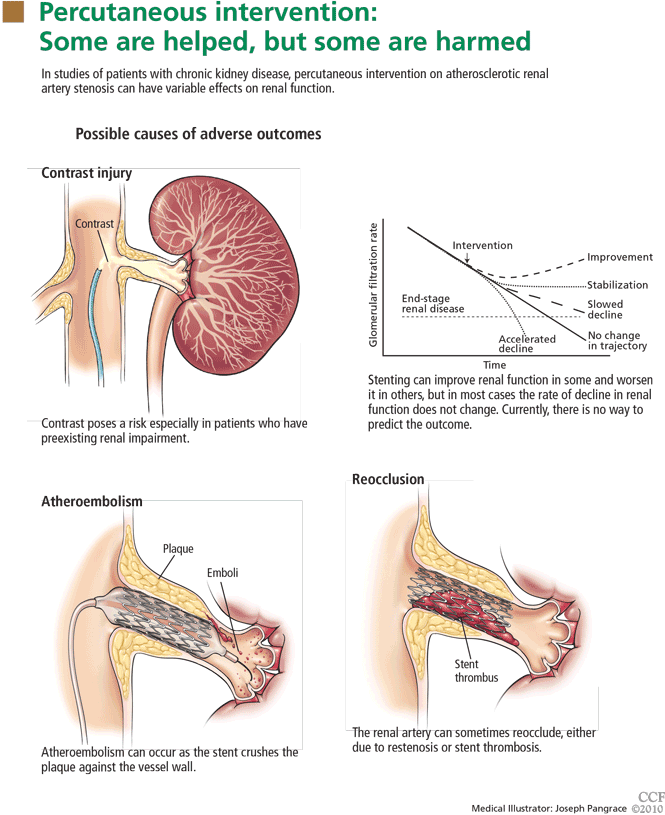
In a prospective cohort study in 304 patients with chronic kidney disease and renal artery stenosis who underwent surgical revascularization, Textor4 reported that the serum creatinine level showed a meaningful improvement afterward in 28%, worsened in 19.7%, and remained unchanged in 160 52.6%. (A “meaningful” change was defined as > 1.0 mg/dL.) Findings were similar in a cohort that underwent stenting.33
Davies et al41 found that 20% of patients who underwent renal stenting had a persistent increase in serum creatinine of 0.5 mg/dL or more. These patients were nearly three times more likely (19% vs 7%) to eventually require dialysis, and they had a lower 5-year survival rate (41% vs 71%).
Zeller et al39 found that renal function improved slightly in 52% of patients who received stents. The mean decrease in serum creatinine in this group was 0.22 mg/dL. However, the other 48% had a mean increase in serum creatinine of 1.1 mg/dL.
From these data we can conclude that, in an unselected population with renal artery stenosis, stenting provides no benefit to renal function, and that the risk of a worsening of renal function after intervention is roughly equal to the likelihood of achieving any benefit.
Other predictors of improvement in renal function have been proposed, but the evidence supporting them has not been consistent. For example, although Radermacher et al42 reported that a renal resistive index (which reflects arterial stiffness downstream of the stenosis) lower than 0.8 predicted a response in renal function, this finding has not been reliably reproduced.43,44 Similarly, while several studies suggest that patients with milder renal dysfunction have a higher likelihood of a renal response,45,46 other studies suggest either that the opposite is true39 or that baseline renal function alone has no impact on outcome.47
In addition, once significant renal atrophy occurs, revascularization may not help much, since irreversible sclerosis has developed. Thus, the goal is to identify kidneys being harmed by renal artery stenosis during the ischemic phase, when the tissue is still viable.
Unfortunately, we still lack a good renal stress test—eg, analogous to the cardiac stress test—to diagnose reversible ischemia in the kidney. The captopril renal scan has that capability but is not accurate in patients with bilateral stenosis or a GFR less than 50 mL/min, severely limiting its applicability.26 Newer technologies such as blood-oxygen-level-dependent (BOLD) magnetic resonance imaging are being investigated for such a role.48
Cohort studies in patients with declining renal function
In several case series, patients whose renal function had been declining before intervention had impressive rates of better renal function afterward.33,39,47,49–54 In a prospective cohort study by Muray et al,47 a rise in serum creatinine of more than 0.1 mg/mL/month before intervention seemed to predict an improvement in renal function afterward.
One would expect that, for renal function to respond to intervention, severe bilateral stenosis or unilateral stenosis to a solitary functioning kidney would need to be present. However, this was an inconsistent finding in these case series.33,39,47,52,53 The Angioplasty and Stent for Renal Artery Lesions (ASTRAL) trial,6,7 discussed later, sheds a bit more light on this.
Stenting usually improves flash pulmonary edema
Acute pulmonary edema in the setting of bilateral renal artery stenosis seems to be a unique case in which improvement in clinical status can be expected in most patients after intervention. Blood pressure improves in 94% to 100% of patients,28,31 renal function either improves or stabilizes in 77% to 91%,28–31 and pulmonary edema resolves without recurrence in 77% to 100%.28–30
NEW RANDOMIZED TRIALS: STAR, ASTRAL, AND CORAL
Despite the lack of evidence supporting revascularization of renal artery stenosis, many interventionalists practice under the assumption that the radiographic finding of renal artery stenosis alone is an indication for renal revascularization. Only three randomized controlled trials in the modern era attempt to examine this hypothesis: STAR, ASTRAL, and CORAL.
STAR: No clear benefit
The Stent Placement and Blood Pressure and Lipid-lowering for the Prevention of Progression of Renal Dysfunction Caused by Atherosclerotic Ostial Stenosis of the Renal Artery (STAR) trial5 was a European multicenter trial that enrolled 140 patients with ostial renal artery stenosis greater than 50%, blood pressure controlled to less than 140/90 mm Hg, and creatinine clearance 15 to 80 mL/min.
Patients were randomized to undergo stenting or medical therapy alone. High blood pressure was treated according to a protocol in which angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers were relegated to second-line use. All patients received a statin, regardless of lipid levels.
At 2 years, the primary end point (a decline in creatinine clearance of 20% or greater) had been reached in 10 (16%) of the 64 patients in the stent group and 16 (22%) of the 76 patients in the medication group; the difference was not statistically significant (hazard ratio 0.73, 95% confidence interval 0.33–1.61). No difference was seen in the secondary end points of the degree of blood pressure control or the rates of cardiovascular morbidity and death.5
ASTRAL: Also no clear benefit
In the international, multicenter ASTRAL trial,6,7 806 patients with at least one stenotic renal artery considered suitable for balloon angioplasty, stenting, or both7 were randomized to undergo intervention or medical management. Hypertension treatment was not specified by a protocol. The mean estimated GFR was 40 mL/min. Most patients (95%–96%) were on statin therapy. The primary outcome was the rate of decline of renal function over time. Secondary outcomes included blood pressure control, renal events, cardiovascular events, and death.
Results. At a mean follow-up of 33.6 months (range 1–4 years), no difference was noted between treatment groups in decline in renal function or blood pressure control at 1 year. Renal function worsened slightly in both groups.
The decline in renal function over time, calculated as the mean slope of the reciprocal of the serum creatinine level over time, was slightly slower in the revascularization group, but the difference was not statistically significant (−0.07 × 10−3 vs −0.13 × 10−3 L/μmol/year, P = .06). This difference did not appear until the last year of the study. There was no difference in the number of patients whose renal function improved or declined during the study.
There was no difference in the rate of any secondary outcome. The medical management group required a slightly higher number of antihypertensive drugs, reaching statistical but not clinical significance (2.97 vs 2.77 drugs, P = .03). More people in the revascularization group were taking ACE inhibitors or angiotensin receptor blockers. There was no difference in the number of patients on any antihypertensive therapy (97% vs 99%). Interestingly, amputations were more common in the revascularization group, occurring in 42 (12%) of the 386 patients in the revascularization group vs 29 (7%) of the 395 patients in the medical group (P = .04).
Seventeen percent of patients randomized to intervention did not have the procedure done. An as-treated analysis of the 317 (83%) patients randomized to revascularization who did receive it showed no differences in outcomes.
There were no differences in outcomes among specific, predefined subgroups based on severity of stenosis at baseline, renal length, baseline estimated GFR, baseline serum creatinine, and rate of progression of renal dysfunction before randomization.7
Comments. ASTRAL contradicts previous nonrandomized studies that suggested that rapidly declining renal function (loss of 20% in 1 year) predicts response to intervention. Considering the large number of patients with unilateral disease in the study, it would be interesting to see what effect stenting had on patients with both severe disease and declining renal function.
ASTRAL has been criticized because it lacked a central laboratory to interpret the severity of stenosis, it did not use a standardized intervention technique (5% of patients underwent angioplasty without stents, although this did not affect outcomes7), and patients were enrolled only if the clinician involved in the case was uncertain of the appropriate management.
This last issue raises the concern for selection bias toward inclusion of more difficult cases that may not respond to intervention. But these shortcomings are not serious enough to negate the fact that preliminary results from the largest randomized controlled trial to date confirm conclusions of other randomized trials, ie, that intervention in renal artery stenosis yields no benefits over medical management in most patients.
Based on the results of STAR and ASTRAL, the practice of indiscriminately revascularizing stenosed renal arteries without strong evidence that the procedure will provide a clinical benefit is no longer tenable. The challenge is to identify those few patients who will respond, and to intervene only on them. Unfortunately, none of the subgroups from ASTRAL helped characterize this population.
CORAL: A large trial is ongoing
The Cardiovascular Outcomes in Renal Artherosclerotic Lesions (CORAL) trial,8 an ongoing multicenter randomized controlled trial in the United States, may be of additional help.
Unlike ASTRAL, CORAL is studying patients who have difficult-to-control hypertension (systolic blood pressure ≥ 155 mm Hg on two or more drugs).8 Chronic kidney disease is not an exclusion criterion unless the serum creatinine concentration is greater than 3.0 mg/dL.
CORAL is using a standardized medical protocol to control blood pressure. In addition, use of embolic protection devices during stenting is encouraged. Hopefully, the large size (a goal of 1,080 patients) and the inclusion of patients with more marked hypertension will address the utility of intervention in higher-risk populations with renal artery stenosis.
RECOMMENDED APPROACH TO INTERVENTION IN RENAL ARTERY STENOSIS
As we wait for CORAL to be completed, we have two modern-era randomized controlled trials that leave us with fewer indications for renal intervention. Table 2 lists commonly cited indications for intervention in renal artery stenosis and the evidence to support them. As most of these are based on retrospective data or have conflicting support in the literature, their utility remains in question. At this point we can safely recommend:
- Patients with preserved or even decreased but stable renal function will not likely have a benefit in renal function after intervention.
- Patients with resistant hypertension may benefit.
- The best evidence supporting intervention is for bilateral stenosis with flash pulmonary edema, but the evidence is from retrospective studies.
- Stenting in bilateral disease without another indication has no apparent benefit.
- Declining renal function is not a guarantee of success.
- It is unclear if patients with severe bilateral stenosis or severe stenosis to a solitary functioning kidney with declining renal function will benefit. Anecdotally, they do respond more often, but as with many other indications for intervention that have gone by the wayside, this may not bear out when studied properly.

As the utility of intervention narrows, the scope of practice for such interventions should narrow accordingly. Attention should now be focusing on clinical, rather than radiographic, indications for intervening on renal artery stenosis.
Therefore, the decision to intervene must not be made solely by the interventionalist. A multidisciplinary approach should be adopted that at the very least includes the input of a nephrologist well versed in renal artery stenosis. In this way, the clinical risks and benefits of renal intervention can be discussed with the patient by providers who are likely to be involved in their care should renal function or hypertension fail to improve afterward.
RISK OF ATHEROEMBOLISM
While renal stenting yields improved technical success in the treatment of renal artery stenosis, it carries with it an increasingly common risk to kidney function: atheroembolism as the stent crushes the plaque against the vessel wall. This may lead to obstruction of the renal microvasculature, increasing the risk of irreversible damage to renal function.
Atheroembolic kidney disease can manifest as progressive renal failure occurring over weeks to months, commonly misdiagnosed as permanent damage from contrast nephropathy.55
Embolic protection devices, inserted downstream of the lesion before stenting to catch any debris that may break loose, have been developed to help address this problem.
Holden et al 57 prospectively studied 63 patients with renal artery stenosis and deteriorating renal function (undefined) who underwent stenting with an embolic protection device. At 6 months after the intervention, renal function had either improved or stabilized in 97% of patients, suggesting that many of the deleterious effects of stenting on renal function are related to atheroembolism.
The Prospective Randomized Study Comparing Renal Artery Stenting With or Without Distal Protection (RESIST) trial, in which renal dysfunction was mild and the GFR was not declining (average estimated GFR 59.3 mL/min), found contrary results.57 In a two-by-two factorial study, patients were randomized to undergo stenting alone, stenting with the antiplatelet agent abciximab (ReoPro), stenting with an embolic protection device, or stenting with both abciximab and an embolic protection device. Interestingly, renal function declined in the first three groups, but remained stable in the group that received both abciximab and an embolic protection device.
ANTIPLATELET THERAPY AFTER RENAL STENTING: HOW LONG?
We have no data on the optimal duration of antiplatelet therapy after renal stenting, and guidelines from professional societies do not comment on it.58 As a result, practice patterns vary significantly among practitioners.
While in-stent restenosis rates are acceptably low after renal stenting, the risks and side effects of antiplatelet therapy often lead to arbitrary withdrawal of these drugs. The effect on stent patency is yet to be determined.
FUTURE DEVELOPMENTS
Results of STAR and ASTRAL confirm the growing suspicion that the surge seen in the last decade in renal artery stenting should be coming to an end. We await results either from CORAL or possibly a post hoc analysis of ASTRAL that might identify potential high-risk groups that will benefit from renal intervention. And as embolic protection devices become more agile and suitable to different renal lesions, there remains the possibility that, due to lower rates of unidentified atheroembolic kidney disease, CORAL may demonstrate improved renal outcomes after stenting. If not, the search for the best means to predict who should have renal intervention will continue.
We know through experience that stenting does provide great benefits for some patients with renal artery stenosis. Furthermore, the clinical problem is too intriguing, and too profitable, to die altogether.
- Choncol M, Linas S. Diagnosis and management of ischemic nephropathy. Clin J Am Soc Nephrol 2006; 1:172–181.
- Webster J, Marshall F, Abdalla M, et al Randomised comparison of percutaneous angioplasty vs continued medical therapy for hypertensive patients with atheromatous renal artery stenosis. Scottish and Newcastle Renal Artery Stenosis Collaborative Group. J Hum Hypertens 1998; 12:329–335.
- Plouin PF, Chatellier G, Darne B, Raynaud A. Blood pressure outcome of angioplasty in atherosclerotic renal artery stenosis: a randomized trial. Essai Multicentrique Medicaments vs Angioplastie (EMMA) Study Group. Hypertension 1998; 31:823–829.
- Textor SC. Revascularization in atherosclerotic renal artery disease. Kidney Int 1998; 53:799–811.
- Bax L, Woittiez AJ, Kouwenberg HJ, et al Stent placement in patients with atherosclerotic renal artery stenosis and impaired renal function: a randomized trial. Ann Intern Med 2009; 150:840–848.
- Mistry S, Ives N, Harding J, et al Angioplasty and STent for Renal Artery Lesions (ASTRAL trial): rationale, methods and results so far. J Hum Hypertens 2007; 21:511–515.
- Wheatley K, Ives N, Kalra P, Moss J. Revascularization versus medical therapy for renal-artery stenosis (ASTRAL). N Engl J Med 2009; 361:1953–1962.
- Cooper CJ, Murphy TP, Matsumoto A, et al Stent revascularization for the prevention of cardiovascular and renal events among patients with renal artery stenosis and systolic hypertension: rationale and design of the CORAL trial. Am Heart J 2006; 152:59–66.
- Galaria II, Surowiec SM, Rhodes JM, et al Percutaneous and open renal revascularizations have equivalent long-term functional outcomes. Ann Vasc Surg 2005; 19:218–228.
- Murphy TP, Soares G, Kim M. Increase in utilization of percutaneous renal artery interventions by Medicare beneficiaries 1996–2000. AJR Am J Roentgenol 2004; 183:561–568.
- Textor SC. Atherosclerotic renal artery stenosis: overtreated but underrated? J Am Soc Nephrol 2008; 19:656–659.
- Kalra PA, Guo H, Gilbertson DT, et al Atherosclerotic renovascular disease in the United States. Kidney Int 2010; 77:37–43.
- Hansen KJ, Edwards MS, Craven TE, et al Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002; 36:443–451.
- Cohen MG, Pascua JA, Garcia-Ben M, et al A simple prediction rule for significant renal artery stenosis in patients undergoing cardiac catheterization. Am Heart J 2005; 150:1204–1211.
- Buller CE, Nogareda JG, Ramanathan K, et al The profile of cardiac patients with renal artery stenosis. J Am Coll Cardiol 2004; 43:1606–1613.
- White CJ, Olin JW. Diagnosis and management of atherosclerotic renal artery stenosis: improving patient selection and outcomes. Nat Clin Pract Cardiovasc Med 2009; 6:176–190.
- Kalra PA, Guo H, Kausz AT, et al Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005; 69:293–301.
- Holley KE, Hunt JC, Brown AL, Kincaid OW, Sheps SG. Renal artery stenosis. A clinical-pathologic study in normotensive and hypertensive patients. Am J Med 1964; 37:14–22.
- de Mast Q, Beutler JJ. The prevalence of atherosclerotic renal artery stenosis in risk groups: a systemic literature review. J Hypertens 2009; 27:1333–1340.
- Kuczera P, Włoszczynska E, Adamczak M, Pencak P, Chudek J, Wiecek A. Frequency of renal artery stenosis and variants of renal vascularization in hypertensive patients: analysis of 1550 angiographies in one centre. J Hum Hypertens 2009; 23:396–401.
- Caps MT, Perissinotto C, Zierler RE, et al Prospective study of atherosclerotic disease progression in the renal artery. Circulation 1998; 98:2866–2872.
- Zierler RE, Bergelin RO, Davidson RC, Cantwell-Gab K, Polissar NL, Strandness DE. A prospective study of disease progression in patients with atherosclerotic renal artery stenosis. Am J Hypertens 1996; 9:1055–1061.
- Schreiber MJ, Pohl MA, Novick AC. The natural history of atherosclerotic and fibrous renal artery disease. Urol Clin North Am 1984; 11:383–392.
- Crowley JJ, Santos RM, Peter RH, et al Progression of renal artery stenosis in patients undergoing cardiac catheterization. Am Heart J 1998; 136:913–918.
- Textor SC. Renovascular hypertension update. Curr Hypertens Rep 2006; 8:521–527.
- Textor SC. Ischemic nephropathy: where are we now? J Am Soc Nephrol 2004; 15:1974–1982.
- Wright JR, Shurrab AE, Cheung C, et al A prospective study of the determinants of renal functional outcome and mortality in atherosclerotic renovascular disease. Am J Kidney Dis 2002; 39:1153–1161.
- Messina LM, Zelenock GB, Yao KA, Stanley JC. Renal revascularization for recurrent pulmonary edema in patients with poorly controlled hypertension and renal insufficiency: a distinct subgroup of patients with arteriosclerotic renal artery occlusive disease. J Vasc Surg 1992; 15:73–80.
- Bloch MJ, Trost DW, Pickering TG, Sos TA, August P. Prevention of recurrent pulmonary edema in patients with bilateral renovascular disease through renal artery stent placement. Am J Hypertens 1999; 12:1–7.
- Gray BH, Olin JW, Childs MB, Sullivan TM, Bacharach JM. Clinical benefit of renal artery angioplasty with stenting for the control of recurrent and refractory congestive heart failure. Vasc Med 2002; 7:275–279.
- Pickering TG, Herman L, Devereux RB, et al Recurrent pulmonary oedema in hypertension due to bilateral renal artery stenosis: treatment by angioplasty or surgical revascularisation. Lancet 1988; 2:551–552.
- Kennedy DJ, Colyer WR, Brewster PS, et al Renal insufficiency as a predictor of adverse events and mortality after renal artery stent placement. Am J Kidney Dis 2003; 14:926–935.
- Beutler JJ, Van Ampting JM, Van De Ven PJ, et al Long-term effects of arterial stenting on kidney function for patients with ostial atherosclerotic renal artery stenosis and renal insufficiency. J Am Soc Nephrol 2001; 12:1475–1481.
- Van de Ven PJ, Kaatee R, Beutler JJ, et al Arterial stenting and balloon angioplasty in ostial atherosclerotic renovascular disease: a randomized trial. Lancet 1999; 353:282–286.
- Isles C, Main J, O’Connell J, et al Survival associated with renovascular disease in Glasgow and Newcastle: a collaborative study. Scott Med J 1990; 35:70–73.
- Hunt JC, Sheps SG, Harrison EG, Strong CG, Bernatz PE. Renal and renovascular hypertension. A reasoned approach to diagnosis and management. Arch Intern Med 1974; 133:988–999.
- Textor SC. Atherosclerotic renal artery stenosis: how big is the problem, and what happens if nothing is done? J Hypertens Suppl 2005; 23:S5–S13.
- van Jaarsveld BC, Krijnen P, Pieterman H, et al The effect of balloon angioplasty on hypertension in atherosclerotic renal-artery stenosis. Dutch Renal Artery Stenosis Intervention Cooperative Study Group. N Engl J Med 2000; 342:1007–1014.
- Zeller T, Frank U, Müller C, et al Predictors of improved renal function after percutaneous stent-supported angioplasty of severe atherosclerotic ostial renal artery stenosis. Circulation 2003; 108;2244–2249.
- Chábová V, Schirger A, Stanson AW, McKusick MA, Textor SC. Outcomes of atherosclerotic renal artery stenosis managed without revascularization. Mayo Clin Proc 2000; 75:437–444.
- Davies MG, Saad WE, Peden EK, Mohiuddin IT, Naoum JJ, Lumsden AB. Implications of acute functional injury following percutaneous renal artery intervention. Ann Vasc Surg 2008; 22:783–789.
- Radermacher J, Chavin A, Bleck J, et al Use of Doppler ultrasonography to predict the outcome of therapy for renal-artery stenosis. N Eng J Med 2001; 344:410–417.
- García-Criado A, Gilabert R, Nicolau C, et al Value of Doppler sonography for predicting clinical outcome after renal artery revascularization in atherosclerotic renal artery stenosis. J Ultrasound Med 2005; 24:1641–1647.
- Zeller T, Müller C, Frank U, et al Stent angioplasty of severe atherosclerotic ostial renal artery stenosis in patients with diabetes mellitus and nephrosclerosis. Catheter Cardiovasc Interv 2003; 58:510–515.
- Harden PN, MacLeod MJ, Rodger RSC, et al Effect of renal-artery stenting on progression of renovascular renal failure. Lancet 1997; 349:1133–1136.
- Isles CG, Robertson S, Hill D. Management of renovascular disease: a review of renal artery stenting in ten studies. QJM 1999; 92:159–167.
- Muray S, Martın M, Amoedo ML, et al Rapid decline in renal function reflects reversibility and predicts the outcome after angioplasty in renal artery stenosis. Am J Kidney Dis 2002; 39:60–66.
- Textor SC, Glockner JF, Lerman LO, et al The use of magnetic resonance to evaluate tissue oxygenation in renal artery stenosis. J Am Soc Nephrol 2008; 19:780–788.
- Paraskevas KI, Perrea D, Briana DD, Liapis CD. Management of atherosclerotic renovascular disease: the effect of renal artery stenting on renal function and blood pressure. Int Urol Nephrol 2006; 38:683–691.
- Watson PS, Hadjipetrou P, Cox SV, Piemonte TC, Eisenhauer AC. Effect of renal artery stenting on renal function and size in patients with atherosclerotic renovascular disease. Circulation 2000; 102:1671–1677.
- Dean RH, Kieffer RW, Smith BM, et al Renovascular hypertension: anatomic and renal function changes during drug therapy. Arch Surg 1981; 116:1408–1415.
- Zhang Q, Shen W, Zhang R, Zhang J, Hu J, Zhang X. Effects of renal artery stenting on renal function and blood pressure in patients with atherosclerotic renovascular disease. Chin Med J (Engl) 2003; 116:1451–1454.
- Ramos F, Kotliar C, Alvarez D, et al Renal function and outcome of PTRA and stenting for atherosclerotic renal artery stenosis. Kidney Int 2003; 63:276–282.
- Rocha-Singh KJ, Ahuja RK, Sung CH, Rutherford J. Long-term renal function preservation after renal artery stenting in patients with progressive ischemic nephropathy. Catheter Cardiovasc Interv 2002; 57:135–141.
- Thadhani RI, Camargo CA, Xavier RJ, Fang LS, Bazari H. Atheroembolic renal failure after invasive procedures. Natural history based on 52 histologically proven cases. Medicine (Baltimore) 1995; 74:350–358.
- Holden A, Hill A, Jaff MR, Pilmore H. Renal artery stent revascularization with embolic protection in patients with ischemic nephropathy. Kidney Int 2006; 70:948–955.
- Cooper CJ, Haller ST, Colyer W, et al Embolic protection and platelet inhibition during renal artery stenting. Circulation 2008; 117:2752–2760.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al ACC/AHA 2005 Practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006; 113:e463–e654.
Author’s note: Atherosclerosis accounts for about 90% of cases of renal artery stenosis in people over age 40.1 Fibromuscular dysplasia, the other major cause, is a separate topic; in this paper “renal artery stenosis” refers to atherosclerotic disease only.
Renal artery stenosis is very common, and the number of angioplasty-stenting procedures performed every year is on the rise. Yet there is no overwhelming evidence that intervention yields clinical benefits—ie, better blood pressure control or renal function— than does medical therapy.
Earlier randomized controlled trials comparing angioplasty without stents and medical management showed no impressive difference in blood pressure.2,3 The data on renal function were even more questionable, with some studies suggesting that, with stenting, the chance of worsening renal function is equal to that of improvement.4
Two large randomized trials comparing renal intervention with medical therapy failed to show any benefit of intervention.5–7 A third study is under way.8
It is time to strongly reconsider the current aggressive approach to revascularization of stenotic renal arteries and take a more coordinated, critical approach.
RENAL INTERVENTIONS ON THE RISE
Renal angioplasty began replacing surgical revascularization in the 1990s, as this less-invasive procedure became more readily available and was shown to have similar clinical outcomes.9 In the last decade, stent placement during angioplasty has become standard, improving the rates of technical success.
As these procedures have become easier to perform and their radiographic outcomes have become more consistent, interventionalists have become more likely, if they see stenosis in a renal artery, to intervene and insert a stent, regardless of proven benefit. In addition, interventionalists from at least three different specialties now compete for these procedures, often by looking at the renal arteries during angiography of other vascular beds (the “drive-by”).
As a result, the number of renal interventions has been rising. Medicare received 21,660 claims for renal artery interventions (surgery or angioplasty) in 2000, compared with 13,380 in 1996—an increase of 62%. However, the number of surgeries actually decreased by 45% during this time, while the number of percutaneous procedures increased by 240%. The number of endovascular claims for renal artery stenosis by cardiologists alone rose 390%.10 Since then, the reports on intervention have been mixed, with one report citing a continued increase in 2005 to 35,000 claims,11 and another suggesting a decrease back to 1997 levels.12
HOW COMMON IS RENAL ARTERY STENOSIS?
The prevalence of renal artery stenosis depends on the definition used and the population screened. It is more common in older patients who have risk factors for other vascular diseases than in the general population.
Renal Doppler ultrasonography can detect stenosis only if the artery is narrowed by more than 60%. Hansen et al13 used ultrasonography to screen 870 people over age 65 and found a lesion (a narrowing of more than 60%) in 6.8%.
Angiography (direct, computed tomographic, or magnetic resonance) can detect less-severe stenosis. Thus, most angiographic studies define renal artery stenosis as a narrowing of more than 50%, and severe disease as a narrowing of more than 70%. Many experts believe that unilateral stenosis needs to be more than 70% to pose a risk to the kidney.14,15
Angiographic prevalence studies have been performed only in patients who were undergoing angiography for another reason such as coronary or peripheral arterial disease that inherently places them at higher risk of renal artery stenosis. For instance, renal artery stenosis is found in 11% to 28% of patients undergoing diagnostic cardiac catheterization. 16
No studies of the prevalence of renal artery stenosis have been performed in the general population. Medicare data indicate that from 1999 to 2001 the incidence of diagnosed renal artery stenosis was 3.7 per 1,000 patientyears. 17 Holley et al,18 in an autopsy series, found renal artery stenosis of greater than 50% in 27% of patients over age 50 and in 56.4% of hypertensive patients. The prevalence was 10% in normotensive patients.
WHO IS AT RISK?
Factors associated with a higher risk of finding renal artery stenosis on a radiographic study include14:
- Older age
- Female sex
- Hypertension
- Three-vessel coronary artery disease
- Peripheral artery disease
- Chronic kidney disease
- Diabetes
- A low level of high-density lipoprotein cholesterol
- The use of at least two cardiovascular drugs.
The prevalence of renal artery stenosis in at-risk populations ranges from 3% to 75% (Table 1).2,4,6,19,20
HOW OFTEN DOES STENOSIS PROGRESS?
The reported rates of progression of atherosclerotic renal artery lesions vary depending on the type of imaging test used and the reason for doing it.
In studies that used duplex ultrasonography, roughly half of lesions smaller than 60% grew to greater than 60% over 3 years.21,22 The risk of total occlusion of an artery was relatively low and depended on the severity of stenosis: 0.7% if the baseline stenosis was less than 60% and 2.3% to 7% if it was greater.21,22
In a seminal study in 1984, Schreiber and colleagues23 compared serial angiograms obtained a mean of 52 months apart in 85 patients who did not undergo intervention. Stenosis had progressed in 37 (44%), and to the point of total occlusion in 14 (16%). In contrast, a 1998 study found progression in 11.1% over 2.6 years, with older patients, women, and those with baseline coronary artery disease at higher risk.24
The the rates of progression differed in these two studies probably because the indications for screening were different (clinical suspicion23 vs routine screening during coronary angiography24), as was the severity of stenosis at the time of diagnosis. Also, when these studies were done, fewer people were taking statins. Thus, similar studies, if repeated, might show even lower rates of progression.
Finally, progression of renal artery stenosis has not been correlated with worsening renal function.
FOUR CLINICAL PRESENTATIONS OF RENAL ARTERY STENOSIS
Renal artery stenosis can present in one of four ways:
Clinically silent stenosis. Because renal artery stenosis is most often found in older patients, who are more likely to have essential hypertension and chronic kidney disease due to other causes, it can be an incidental finding that is completely clinically silent.16,25
Renovascular hypertension is defined as high blood pressure due to up-regulation of neurohormonal activity in response to decreased perfusion from renal artery stenosis. Renal artery stenosis is estimated to be the cause of hypertension in only 0.5% to 4.0% of hypertensive patients, or in 26% of patients with secondary hypertension.3
Ischemic nephropathy is more difficult to define because ischemia alone rarely explains the damage done to the kidneys. Activation of neurohormonal pathways and microvascular injury are thought to contribute to oxidative stress and fibrosis.26 These phenomena may explain why similar degrees of stenosis lead to varying degrees of kidney damage in different patients and why the severity of stenosis does not correlate with the degree of renal dysfunction.27
Furthermore, stenosis may lead to irreversible but stable kidney damage. It is therefore not surprising that, in studies in unselected populations (ie, studies that included patients with all presentations of renal artery stenosis, not just those more likely to benefit), up to two-thirds of renal interventions yielded no clinical benefit.25
As a result, if we define ischemic nephropathy as renal artery stenosis with renal dysfunction not attributable to another cause, we probably will overestimate the prevalence of ischemic nephropathy, leading to overly optimistic expectations about the response to revascularization.
Recurrent “flash” pulmonary edema is a less common presentation, usually occurring in patients with critical bilateral renal artery stenosis or unilateral stenosis in an artery supplying a solitary functioning kidney. Most have severe hypertension (average systolic blood pressure 174–207 mm Hg) and poor renal function.28–30
The association between pulmonary edema and bilateral renal artery stenosis was first noted in 1998 by Pickering et al,31 who in several case series showed that 82% to 92% of patients with recurrent pulmonary edema and renal artery stenosis had bilateral stenosis, compared with 20% to 65% of those with other presentations. Later case series corroborated this finding: 85% to 100% of patients with renal artery stenosis and pulmonary edema had bilateral stenosis.28–30
STENTING IS NOW STANDARD
Stenting has become standard in the endovascular treatment of renal artery stenosis.
Most atherosclerotic renal artery lesions are located in the ostium (ie, where the artery branches off from the aorta), and many are extensions of calcified aortic plaque.26,32 These hard lesions tend to rebound to their original shape more often with balloon angioplasty alone. Stenting provides the additional force needed to permanently disrupt the lesion, leading to a longer-lasting result.
Rates of technical success (dilating the artery during the intervention) are higher with stents than without them (98% vs 46%– 77%).33,34 If the lesion is ostial, this difference is even more impressive (75% vs 29%). In addition, restenosis rates at 6 months are lower with stents (14% vs 26%–48%).34
GOALS: LOWER THE BLOOD PRESSURE, SAVE THE KIDNEY
Because endovascular procedures pose some risk to the patient, it is critical to intervene only in patients most likely to respond clinically. The decision to intervene depends largely on the clinical goal, which should depend on the clinical presentation.
However, if renal artery stenosis is clinically silent, most of the evidence suggests that intervention has no benefit. Furthermore, although retrospective studies have indicated that intervention may improve survival rates,35,36 prospective studies have not. Similarly, studies have not shown that intervention generally improves cardiovascular outcomes, even though renal artery stenosis is associated with cardiovascular risk.
Hypertension plus stenosis is not necessarily renovascular hypertension
Essential hypertension and clinically silent renal artery stenosis often coexist, which is why blood pressure control often does not improve after stenting. Also, essential hypertension often coexists with renovascular hypertension.37 In this situation, stenting may not eliminate the need for antihypertensive drugs, although it may improve blood pressure control and decrease the drug burden.
Before stents came into use, several randomized controlled trials found that blood pressure was no better controlled after angioplasty, 2,3,38 except in cases of bilateral stenosis.2 This may be because stenosis tended to recur after angioplasty without stents.
The 2000 Dutch Renal Artery Stenosis Intervention Cooperative (DRASTIC) study was the first randomized controlled trial to examine the effect of angioplasty on blood pressure control in renal artery stenosis.38 It had significant design flaws. For example, many patients crossed over from the medical management group to the intervention group because their hypertension was resistant to medical therapy. Overall, intervention (balloon angioplasty without stents in 54 of 56 patients, with stents in the other 2) carried no benefit. However, in subgroup analysis, the patients who crossed over because of resistant hypertension (failure of a three-drug regimen) were more likely to benefit from angioplasty. This suggested that risk stratification should take place early on, before proceeding with revascularization.
With stents, Zeller,39 in a prospective nonrandomized study, found that the mean arterial pressure decreased by 10 mm Hg. Randomized trials (see below) have failed to demonstrate such a benefit.
Stenting may not improve renal function
Coincidental renal artery stenosis in a patient with unrelated chronic kidney disease is very hard to differentiate from true ischemic nephropathy. Furthermore, most patients with ischemic nephropathy do not benefit from revascularization, making it challenging to identify those few whose renal function may respond.
Given that patients with chronic kidney disease tend to have a higher risk of cardiovascular disease, it is not surprising that 15% of them may have renal artery stenosis,4 most often incidental.
Chábová40 examined the outcomes of 68 patients who had chronic kidney disease and a renal artery lesion larger than 70% who did not undergo angioplasty. In only 10 (15%) of the patients did the glomerular filtration rate (GFR) decline by more than 50% of its baseline value during the study period of 3 years. Given the retrospective nature of the study, it cannot be determined (and is rather unlikely) that ischemic nephropathy was the cause of the decline in kidney function in all 10 patients.
In a prospective cohort study in 304 patients with chronic kidney disease and renal artery stenosis who underwent surgical revascularization, Textor4 reported that the serum creatinine level showed a meaningful improvement afterward in 28%, worsened in 19.7%, and remained unchanged in 160 52.6%. (A “meaningful” change was defined as > 1.0 mg/dL.) Findings were similar in a cohort that underwent stenting.33
Davies et al41 found that 20% of patients who underwent renal stenting had a persistent increase in serum creatinine of 0.5 mg/dL or more. These patients were nearly three times more likely (19% vs 7%) to eventually require dialysis, and they had a lower 5-year survival rate (41% vs 71%).
Zeller et al39 found that renal function improved slightly in 52% of patients who received stents. The mean decrease in serum creatinine in this group was 0.22 mg/dL. However, the other 48% had a mean increase in serum creatinine of 1.1 mg/dL.
From these data we can conclude that, in an unselected population with renal artery stenosis, stenting provides no benefit to renal function, and that the risk of a worsening of renal function after intervention is roughly equal to the likelihood of achieving any benefit.
Other predictors of improvement in renal function have been proposed, but the evidence supporting them has not been consistent. For example, although Radermacher et al42 reported that a renal resistive index (which reflects arterial stiffness downstream of the stenosis) lower than 0.8 predicted a response in renal function, this finding has not been reliably reproduced.43,44 Similarly, while several studies suggest that patients with milder renal dysfunction have a higher likelihood of a renal response,45,46 other studies suggest either that the opposite is true39 or that baseline renal function alone has no impact on outcome.47
In addition, once significant renal atrophy occurs, revascularization may not help much, since irreversible sclerosis has developed. Thus, the goal is to identify kidneys being harmed by renal artery stenosis during the ischemic phase, when the tissue is still viable.
Unfortunately, we still lack a good renal stress test—eg, analogous to the cardiac stress test—to diagnose reversible ischemia in the kidney. The captopril renal scan has that capability but is not accurate in patients with bilateral stenosis or a GFR less than 50 mL/min, severely limiting its applicability.26 Newer technologies such as blood-oxygen-level-dependent (BOLD) magnetic resonance imaging are being investigated for such a role.48
Cohort studies in patients with declining renal function
In several case series, patients whose renal function had been declining before intervention had impressive rates of better renal function afterward.33,39,47,49–54 In a prospective cohort study by Muray et al,47 a rise in serum creatinine of more than 0.1 mg/mL/month before intervention seemed to predict an improvement in renal function afterward.
One would expect that, for renal function to respond to intervention, severe bilateral stenosis or unilateral stenosis to a solitary functioning kidney would need to be present. However, this was an inconsistent finding in these case series.33,39,47,52,53 The Angioplasty and Stent for Renal Artery Lesions (ASTRAL) trial,6,7 discussed later, sheds a bit more light on this.
Stenting usually improves flash pulmonary edema
Acute pulmonary edema in the setting of bilateral renal artery stenosis seems to be a unique case in which improvement in clinical status can be expected in most patients after intervention. Blood pressure improves in 94% to 100% of patients,28,31 renal function either improves or stabilizes in 77% to 91%,28–31 and pulmonary edema resolves without recurrence in 77% to 100%.28–30
NEW RANDOMIZED TRIALS: STAR, ASTRAL, AND CORAL
Despite the lack of evidence supporting revascularization of renal artery stenosis, many interventionalists practice under the assumption that the radiographic finding of renal artery stenosis alone is an indication for renal revascularization. Only three randomized controlled trials in the modern era attempt to examine this hypothesis: STAR, ASTRAL, and CORAL.
STAR: No clear benefit
The Stent Placement and Blood Pressure and Lipid-lowering for the Prevention of Progression of Renal Dysfunction Caused by Atherosclerotic Ostial Stenosis of the Renal Artery (STAR) trial5 was a European multicenter trial that enrolled 140 patients with ostial renal artery stenosis greater than 50%, blood pressure controlled to less than 140/90 mm Hg, and creatinine clearance 15 to 80 mL/min.
Patients were randomized to undergo stenting or medical therapy alone. High blood pressure was treated according to a protocol in which angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers were relegated to second-line use. All patients received a statin, regardless of lipid levels.
At 2 years, the primary end point (a decline in creatinine clearance of 20% or greater) had been reached in 10 (16%) of the 64 patients in the stent group and 16 (22%) of the 76 patients in the medication group; the difference was not statistically significant (hazard ratio 0.73, 95% confidence interval 0.33–1.61). No difference was seen in the secondary end points of the degree of blood pressure control or the rates of cardiovascular morbidity and death.5
ASTRAL: Also no clear benefit
In the international, multicenter ASTRAL trial,6,7 806 patients with at least one stenotic renal artery considered suitable for balloon angioplasty, stenting, or both7 were randomized to undergo intervention or medical management. Hypertension treatment was not specified by a protocol. The mean estimated GFR was 40 mL/min. Most patients (95%–96%) were on statin therapy. The primary outcome was the rate of decline of renal function over time. Secondary outcomes included blood pressure control, renal events, cardiovascular events, and death.
Results. At a mean follow-up of 33.6 months (range 1–4 years), no difference was noted between treatment groups in decline in renal function or blood pressure control at 1 year. Renal function worsened slightly in both groups.
The decline in renal function over time, calculated as the mean slope of the reciprocal of the serum creatinine level over time, was slightly slower in the revascularization group, but the difference was not statistically significant (−0.07 × 10−3 vs −0.13 × 10−3 L/μmol/year, P = .06). This difference did not appear until the last year of the study. There was no difference in the number of patients whose renal function improved or declined during the study.
There was no difference in the rate of any secondary outcome. The medical management group required a slightly higher number of antihypertensive drugs, reaching statistical but not clinical significance (2.97 vs 2.77 drugs, P = .03). More people in the revascularization group were taking ACE inhibitors or angiotensin receptor blockers. There was no difference in the number of patients on any antihypertensive therapy (97% vs 99%). Interestingly, amputations were more common in the revascularization group, occurring in 42 (12%) of the 386 patients in the revascularization group vs 29 (7%) of the 395 patients in the medical group (P = .04).
Seventeen percent of patients randomized to intervention did not have the procedure done. An as-treated analysis of the 317 (83%) patients randomized to revascularization who did receive it showed no differences in outcomes.
There were no differences in outcomes among specific, predefined subgroups based on severity of stenosis at baseline, renal length, baseline estimated GFR, baseline serum creatinine, and rate of progression of renal dysfunction before randomization.7
Comments. ASTRAL contradicts previous nonrandomized studies that suggested that rapidly declining renal function (loss of 20% in 1 year) predicts response to intervention. Considering the large number of patients with unilateral disease in the study, it would be interesting to see what effect stenting had on patients with both severe disease and declining renal function.
ASTRAL has been criticized because it lacked a central laboratory to interpret the severity of stenosis, it did not use a standardized intervention technique (5% of patients underwent angioplasty without stents, although this did not affect outcomes7), and patients were enrolled only if the clinician involved in the case was uncertain of the appropriate management.
This last issue raises the concern for selection bias toward inclusion of more difficult cases that may not respond to intervention. But these shortcomings are not serious enough to negate the fact that preliminary results from the largest randomized controlled trial to date confirm conclusions of other randomized trials, ie, that intervention in renal artery stenosis yields no benefits over medical management in most patients.
Based on the results of STAR and ASTRAL, the practice of indiscriminately revascularizing stenosed renal arteries without strong evidence that the procedure will provide a clinical benefit is no longer tenable. The challenge is to identify those few patients who will respond, and to intervene only on them. Unfortunately, none of the subgroups from ASTRAL helped characterize this population.
CORAL: A large trial is ongoing
The Cardiovascular Outcomes in Renal Artherosclerotic Lesions (CORAL) trial,8 an ongoing multicenter randomized controlled trial in the United States, may be of additional help.
Unlike ASTRAL, CORAL is studying patients who have difficult-to-control hypertension (systolic blood pressure ≥ 155 mm Hg on two or more drugs).8 Chronic kidney disease is not an exclusion criterion unless the serum creatinine concentration is greater than 3.0 mg/dL.
CORAL is using a standardized medical protocol to control blood pressure. In addition, use of embolic protection devices during stenting is encouraged. Hopefully, the large size (a goal of 1,080 patients) and the inclusion of patients with more marked hypertension will address the utility of intervention in higher-risk populations with renal artery stenosis.
RECOMMENDED APPROACH TO INTERVENTION IN RENAL ARTERY STENOSIS
As we wait for CORAL to be completed, we have two modern-era randomized controlled trials that leave us with fewer indications for renal intervention. Table 2 lists commonly cited indications for intervention in renal artery stenosis and the evidence to support them. As most of these are based on retrospective data or have conflicting support in the literature, their utility remains in question. At this point we can safely recommend:
- Patients with preserved or even decreased but stable renal function will not likely have a benefit in renal function after intervention.
- Patients with resistant hypertension may benefit.
- The best evidence supporting intervention is for bilateral stenosis with flash pulmonary edema, but the evidence is from retrospective studies.
- Stenting in bilateral disease without another indication has no apparent benefit.
- Declining renal function is not a guarantee of success.
- It is unclear if patients with severe bilateral stenosis or severe stenosis to a solitary functioning kidney with declining renal function will benefit. Anecdotally, they do respond more often, but as with many other indications for intervention that have gone by the wayside, this may not bear out when studied properly.
As the utility of intervention narrows, the scope of practice for such interventions should narrow accordingly. Attention should now be focusing on clinical, rather than radiographic, indications for intervening on renal artery stenosis.
Therefore, the decision to intervene must not be made solely by the interventionalist. A multidisciplinary approach should be adopted that at the very least includes the input of a nephrologist well versed in renal artery stenosis. In this way, the clinical risks and benefits of renal intervention can be discussed with the patient by providers who are likely to be involved in their care should renal function or hypertension fail to improve afterward.
RISK OF ATHEROEMBOLISM
While renal stenting yields improved technical success in the treatment of renal artery stenosis, it carries with it an increasingly common risk to kidney function: atheroembolism as the stent crushes the plaque against the vessel wall. This may lead to obstruction of the renal microvasculature, increasing the risk of irreversible damage to renal function.
Atheroembolic kidney disease can manifest as progressive renal failure occurring over weeks to months, commonly misdiagnosed as permanent damage from contrast nephropathy.55
Embolic protection devices, inserted downstream of the lesion before stenting to catch any debris that may break loose, have been developed to help address this problem.
Holden et al 57 prospectively studied 63 patients with renal artery stenosis and deteriorating renal function (undefined) who underwent stenting with an embolic protection device. At 6 months after the intervention, renal function had either improved or stabilized in 97% of patients, suggesting that many of the deleterious effects of stenting on renal function are related to atheroembolism.
The Prospective Randomized Study Comparing Renal Artery Stenting With or Without Distal Protection (RESIST) trial, in which renal dysfunction was mild and the GFR was not declining (average estimated GFR 59.3 mL/min), found contrary results.57 In a two-by-two factorial study, patients were randomized to undergo stenting alone, stenting with the antiplatelet agent abciximab (ReoPro), stenting with an embolic protection device, or stenting with both abciximab and an embolic protection device. Interestingly, renal function declined in the first three groups, but remained stable in the group that received both abciximab and an embolic protection device.
ANTIPLATELET THERAPY AFTER RENAL STENTING: HOW LONG?
We have no data on the optimal duration of antiplatelet therapy after renal stenting, and guidelines from professional societies do not comment on it.58 As a result, practice patterns vary significantly among practitioners.
While in-stent restenosis rates are acceptably low after renal stenting, the risks and side effects of antiplatelet therapy often lead to arbitrary withdrawal of these drugs. The effect on stent patency is yet to be determined.
FUTURE DEVELOPMENTS
Results of STAR and ASTRAL confirm the growing suspicion that the surge seen in the last decade in renal artery stenting should be coming to an end. We await results either from CORAL or possibly a post hoc analysis of ASTRAL that might identify potential high-risk groups that will benefit from renal intervention. And as embolic protection devices become more agile and suitable to different renal lesions, there remains the possibility that, due to lower rates of unidentified atheroembolic kidney disease, CORAL may demonstrate improved renal outcomes after stenting. If not, the search for the best means to predict who should have renal intervention will continue.
We know through experience that stenting does provide great benefits for some patients with renal artery stenosis. Furthermore, the clinical problem is too intriguing, and too profitable, to die altogether.
Author’s note: Atherosclerosis accounts for about 90% of cases of renal artery stenosis in people over age 40.1 Fibromuscular dysplasia, the other major cause, is a separate topic; in this paper “renal artery stenosis” refers to atherosclerotic disease only.
Renal artery stenosis is very common, and the number of angioplasty-stenting procedures performed every year is on the rise. Yet there is no overwhelming evidence that intervention yields clinical benefits—ie, better blood pressure control or renal function— than does medical therapy.
Earlier randomized controlled trials comparing angioplasty without stents and medical management showed no impressive difference in blood pressure.2,3 The data on renal function were even more questionable, with some studies suggesting that, with stenting, the chance of worsening renal function is equal to that of improvement.4
Two large randomized trials comparing renal intervention with medical therapy failed to show any benefit of intervention.5–7 A third study is under way.8
It is time to strongly reconsider the current aggressive approach to revascularization of stenotic renal arteries and take a more coordinated, critical approach.
RENAL INTERVENTIONS ON THE RISE
Renal angioplasty began replacing surgical revascularization in the 1990s, as this less-invasive procedure became more readily available and was shown to have similar clinical outcomes.9 In the last decade, stent placement during angioplasty has become standard, improving the rates of technical success.
As these procedures have become easier to perform and their radiographic outcomes have become more consistent, interventionalists have become more likely, if they see stenosis in a renal artery, to intervene and insert a stent, regardless of proven benefit. In addition, interventionalists from at least three different specialties now compete for these procedures, often by looking at the renal arteries during angiography of other vascular beds (the “drive-by”).
As a result, the number of renal interventions has been rising. Medicare received 21,660 claims for renal artery interventions (surgery or angioplasty) in 2000, compared with 13,380 in 1996—an increase of 62%. However, the number of surgeries actually decreased by 45% during this time, while the number of percutaneous procedures increased by 240%. The number of endovascular claims for renal artery stenosis by cardiologists alone rose 390%.10 Since then, the reports on intervention have been mixed, with one report citing a continued increase in 2005 to 35,000 claims,11 and another suggesting a decrease back to 1997 levels.12
HOW COMMON IS RENAL ARTERY STENOSIS?
The prevalence of renal artery stenosis depends on the definition used and the population screened. It is more common in older patients who have risk factors for other vascular diseases than in the general population.
Renal Doppler ultrasonography can detect stenosis only if the artery is narrowed by more than 60%. Hansen et al13 used ultrasonography to screen 870 people over age 65 and found a lesion (a narrowing of more than 60%) in 6.8%.
Angiography (direct, computed tomographic, or magnetic resonance) can detect less-severe stenosis. Thus, most angiographic studies define renal artery stenosis as a narrowing of more than 50%, and severe disease as a narrowing of more than 70%. Many experts believe that unilateral stenosis needs to be more than 70% to pose a risk to the kidney.14,15
Angiographic prevalence studies have been performed only in patients who were undergoing angiography for another reason such as coronary or peripheral arterial disease that inherently places them at higher risk of renal artery stenosis. For instance, renal artery stenosis is found in 11% to 28% of patients undergoing diagnostic cardiac catheterization. 16
No studies of the prevalence of renal artery stenosis have been performed in the general population. Medicare data indicate that from 1999 to 2001 the incidence of diagnosed renal artery stenosis was 3.7 per 1,000 patientyears. 17 Holley et al,18 in an autopsy series, found renal artery stenosis of greater than 50% in 27% of patients over age 50 and in 56.4% of hypertensive patients. The prevalence was 10% in normotensive patients.
WHO IS AT RISK?
Factors associated with a higher risk of finding renal artery stenosis on a radiographic study include14:
- Older age
- Female sex
- Hypertension
- Three-vessel coronary artery disease
- Peripheral artery disease
- Chronic kidney disease
- Diabetes
- A low level of high-density lipoprotein cholesterol
- The use of at least two cardiovascular drugs.
The prevalence of renal artery stenosis in at-risk populations ranges from 3% to 75% (Table 1).2,4,6,19,20
HOW OFTEN DOES STENOSIS PROGRESS?
The reported rates of progression of atherosclerotic renal artery lesions vary depending on the type of imaging test used and the reason for doing it.
In studies that used duplex ultrasonography, roughly half of lesions smaller than 60% grew to greater than 60% over 3 years.21,22 The risk of total occlusion of an artery was relatively low and depended on the severity of stenosis: 0.7% if the baseline stenosis was less than 60% and 2.3% to 7% if it was greater.21,22
In a seminal study in 1984, Schreiber and colleagues23 compared serial angiograms obtained a mean of 52 months apart in 85 patients who did not undergo intervention. Stenosis had progressed in 37 (44%), and to the point of total occlusion in 14 (16%). In contrast, a 1998 study found progression in 11.1% over 2.6 years, with older patients, women, and those with baseline coronary artery disease at higher risk.24
The the rates of progression differed in these two studies probably because the indications for screening were different (clinical suspicion23 vs routine screening during coronary angiography24), as was the severity of stenosis at the time of diagnosis. Also, when these studies were done, fewer people were taking statins. Thus, similar studies, if repeated, might show even lower rates of progression.
Finally, progression of renal artery stenosis has not been correlated with worsening renal function.
FOUR CLINICAL PRESENTATIONS OF RENAL ARTERY STENOSIS
Renal artery stenosis can present in one of four ways:
Clinically silent stenosis. Because renal artery stenosis is most often found in older patients, who are more likely to have essential hypertension and chronic kidney disease due to other causes, it can be an incidental finding that is completely clinically silent.16,25
Renovascular hypertension is defined as high blood pressure due to up-regulation of neurohormonal activity in response to decreased perfusion from renal artery stenosis. Renal artery stenosis is estimated to be the cause of hypertension in only 0.5% to 4.0% of hypertensive patients, or in 26% of patients with secondary hypertension.3
Ischemic nephropathy is more difficult to define because ischemia alone rarely explains the damage done to the kidneys. Activation of neurohormonal pathways and microvascular injury are thought to contribute to oxidative stress and fibrosis.26 These phenomena may explain why similar degrees of stenosis lead to varying degrees of kidney damage in different patients and why the severity of stenosis does not correlate with the degree of renal dysfunction.27
Furthermore, stenosis may lead to irreversible but stable kidney damage. It is therefore not surprising that, in studies in unselected populations (ie, studies that included patients with all presentations of renal artery stenosis, not just those more likely to benefit), up to two-thirds of renal interventions yielded no clinical benefit.25
As a result, if we define ischemic nephropathy as renal artery stenosis with renal dysfunction not attributable to another cause, we probably will overestimate the prevalence of ischemic nephropathy, leading to overly optimistic expectations about the response to revascularization.
Recurrent “flash” pulmonary edema is a less common presentation, usually occurring in patients with critical bilateral renal artery stenosis or unilateral stenosis in an artery supplying a solitary functioning kidney. Most have severe hypertension (average systolic blood pressure 174–207 mm Hg) and poor renal function.28–30
The association between pulmonary edema and bilateral renal artery stenosis was first noted in 1998 by Pickering et al,31 who in several case series showed that 82% to 92% of patients with recurrent pulmonary edema and renal artery stenosis had bilateral stenosis, compared with 20% to 65% of those with other presentations. Later case series corroborated this finding: 85% to 100% of patients with renal artery stenosis and pulmonary edema had bilateral stenosis.28–30
STENTING IS NOW STANDARD
Stenting has become standard in the endovascular treatment of renal artery stenosis.
Most atherosclerotic renal artery lesions are located in the ostium (ie, where the artery branches off from the aorta), and many are extensions of calcified aortic plaque.26,32 These hard lesions tend to rebound to their original shape more often with balloon angioplasty alone. Stenting provides the additional force needed to permanently disrupt the lesion, leading to a longer-lasting result.
Rates of technical success (dilating the artery during the intervention) are higher with stents than without them (98% vs 46%– 77%).33,34 If the lesion is ostial, this difference is even more impressive (75% vs 29%). In addition, restenosis rates at 6 months are lower with stents (14% vs 26%–48%).34
GOALS: LOWER THE BLOOD PRESSURE, SAVE THE KIDNEY
Because endovascular procedures pose some risk to the patient, it is critical to intervene only in patients most likely to respond clinically. The decision to intervene depends largely on the clinical goal, which should depend on the clinical presentation.
However, if renal artery stenosis is clinically silent, most of the evidence suggests that intervention has no benefit. Furthermore, although retrospective studies have indicated that intervention may improve survival rates,35,36 prospective studies have not. Similarly, studies have not shown that intervention generally improves cardiovascular outcomes, even though renal artery stenosis is associated with cardiovascular risk.
Hypertension plus stenosis is not necessarily renovascular hypertension
Essential hypertension and clinically silent renal artery stenosis often coexist, which is why blood pressure control often does not improve after stenting. Also, essential hypertension often coexists with renovascular hypertension.37 In this situation, stenting may not eliminate the need for antihypertensive drugs, although it may improve blood pressure control and decrease the drug burden.
Before stents came into use, several randomized controlled trials found that blood pressure was no better controlled after angioplasty, 2,3,38 except in cases of bilateral stenosis.2 This may be because stenosis tended to recur after angioplasty without stents.
The 2000 Dutch Renal Artery Stenosis Intervention Cooperative (DRASTIC) study was the first randomized controlled trial to examine the effect of angioplasty on blood pressure control in renal artery stenosis.38 It had significant design flaws. For example, many patients crossed over from the medical management group to the intervention group because their hypertension was resistant to medical therapy. Overall, intervention (balloon angioplasty without stents in 54 of 56 patients, with stents in the other 2) carried no benefit. However, in subgroup analysis, the patients who crossed over because of resistant hypertension (failure of a three-drug regimen) were more likely to benefit from angioplasty. This suggested that risk stratification should take place early on, before proceeding with revascularization.
With stents, Zeller,39 in a prospective nonrandomized study, found that the mean arterial pressure decreased by 10 mm Hg. Randomized trials (see below) have failed to demonstrate such a benefit.
Stenting may not improve renal function
Coincidental renal artery stenosis in a patient with unrelated chronic kidney disease is very hard to differentiate from true ischemic nephropathy. Furthermore, most patients with ischemic nephropathy do not benefit from revascularization, making it challenging to identify those few whose renal function may respond.
Given that patients with chronic kidney disease tend to have a higher risk of cardiovascular disease, it is not surprising that 15% of them may have renal artery stenosis,4 most often incidental.
Chábová40 examined the outcomes of 68 patients who had chronic kidney disease and a renal artery lesion larger than 70% who did not undergo angioplasty. In only 10 (15%) of the patients did the glomerular filtration rate (GFR) decline by more than 50% of its baseline value during the study period of 3 years. Given the retrospective nature of the study, it cannot be determined (and is rather unlikely) that ischemic nephropathy was the cause of the decline in kidney function in all 10 patients.
In a prospective cohort study in 304 patients with chronic kidney disease and renal artery stenosis who underwent surgical revascularization, Textor4 reported that the serum creatinine level showed a meaningful improvement afterward in 28%, worsened in 19.7%, and remained unchanged in 160 52.6%. (A “meaningful” change was defined as > 1.0 mg/dL.) Findings were similar in a cohort that underwent stenting.33
Davies et al41 found that 20% of patients who underwent renal stenting had a persistent increase in serum creatinine of 0.5 mg/dL or more. These patients were nearly three times more likely (19% vs 7%) to eventually require dialysis, and they had a lower 5-year survival rate (41% vs 71%).
Zeller et al39 found that renal function improved slightly in 52% of patients who received stents. The mean decrease in serum creatinine in this group was 0.22 mg/dL. However, the other 48% had a mean increase in serum creatinine of 1.1 mg/dL.
From these data we can conclude that, in an unselected population with renal artery stenosis, stenting provides no benefit to renal function, and that the risk of a worsening of renal function after intervention is roughly equal to the likelihood of achieving any benefit.
Other predictors of improvement in renal function have been proposed, but the evidence supporting them has not been consistent. For example, although Radermacher et al42 reported that a renal resistive index (which reflects arterial stiffness downstream of the stenosis) lower than 0.8 predicted a response in renal function, this finding has not been reliably reproduced.43,44 Similarly, while several studies suggest that patients with milder renal dysfunction have a higher likelihood of a renal response,45,46 other studies suggest either that the opposite is true39 or that baseline renal function alone has no impact on outcome.47
In addition, once significant renal atrophy occurs, revascularization may not help much, since irreversible sclerosis has developed. Thus, the goal is to identify kidneys being harmed by renal artery stenosis during the ischemic phase, when the tissue is still viable.
Unfortunately, we still lack a good renal stress test—eg, analogous to the cardiac stress test—to diagnose reversible ischemia in the kidney. The captopril renal scan has that capability but is not accurate in patients with bilateral stenosis or a GFR less than 50 mL/min, severely limiting its applicability.26 Newer technologies such as blood-oxygen-level-dependent (BOLD) magnetic resonance imaging are being investigated for such a role.48
Cohort studies in patients with declining renal function
In several case series, patients whose renal function had been declining before intervention had impressive rates of better renal function afterward.33,39,47,49–54 In a prospective cohort study by Muray et al,47 a rise in serum creatinine of more than 0.1 mg/mL/month before intervention seemed to predict an improvement in renal function afterward.
One would expect that, for renal function to respond to intervention, severe bilateral stenosis or unilateral stenosis to a solitary functioning kidney would need to be present. However, this was an inconsistent finding in these case series.33,39,47,52,53 The Angioplasty and Stent for Renal Artery Lesions (ASTRAL) trial,6,7 discussed later, sheds a bit more light on this.
Stenting usually improves flash pulmonary edema
Acute pulmonary edema in the setting of bilateral renal artery stenosis seems to be a unique case in which improvement in clinical status can be expected in most patients after intervention. Blood pressure improves in 94% to 100% of patients,28,31 renal function either improves or stabilizes in 77% to 91%,28–31 and pulmonary edema resolves without recurrence in 77% to 100%.28–30
NEW RANDOMIZED TRIALS: STAR, ASTRAL, AND CORAL
Despite the lack of evidence supporting revascularization of renal artery stenosis, many interventionalists practice under the assumption that the radiographic finding of renal artery stenosis alone is an indication for renal revascularization. Only three randomized controlled trials in the modern era attempt to examine this hypothesis: STAR, ASTRAL, and CORAL.
STAR: No clear benefit
The Stent Placement and Blood Pressure and Lipid-lowering for the Prevention of Progression of Renal Dysfunction Caused by Atherosclerotic Ostial Stenosis of the Renal Artery (STAR) trial5 was a European multicenter trial that enrolled 140 patients with ostial renal artery stenosis greater than 50%, blood pressure controlled to less than 140/90 mm Hg, and creatinine clearance 15 to 80 mL/min.
Patients were randomized to undergo stenting or medical therapy alone. High blood pressure was treated according to a protocol in which angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers were relegated to second-line use. All patients received a statin, regardless of lipid levels.
At 2 years, the primary end point (a decline in creatinine clearance of 20% or greater) had been reached in 10 (16%) of the 64 patients in the stent group and 16 (22%) of the 76 patients in the medication group; the difference was not statistically significant (hazard ratio 0.73, 95% confidence interval 0.33–1.61). No difference was seen in the secondary end points of the degree of blood pressure control or the rates of cardiovascular morbidity and death.5
ASTRAL: Also no clear benefit
In the international, multicenter ASTRAL trial,6,7 806 patients with at least one stenotic renal artery considered suitable for balloon angioplasty, stenting, or both7 were randomized to undergo intervention or medical management. Hypertension treatment was not specified by a protocol. The mean estimated GFR was 40 mL/min. Most patients (95%–96%) were on statin therapy. The primary outcome was the rate of decline of renal function over time. Secondary outcomes included blood pressure control, renal events, cardiovascular events, and death.
Results. At a mean follow-up of 33.6 months (range 1–4 years), no difference was noted between treatment groups in decline in renal function or blood pressure control at 1 year. Renal function worsened slightly in both groups.
The decline in renal function over time, calculated as the mean slope of the reciprocal of the serum creatinine level over time, was slightly slower in the revascularization group, but the difference was not statistically significant (−0.07 × 10−3 vs −0.13 × 10−3 L/μmol/year, P = .06). This difference did not appear until the last year of the study. There was no difference in the number of patients whose renal function improved or declined during the study.
There was no difference in the rate of any secondary outcome. The medical management group required a slightly higher number of antihypertensive drugs, reaching statistical but not clinical significance (2.97 vs 2.77 drugs, P = .03). More people in the revascularization group were taking ACE inhibitors or angiotensin receptor blockers. There was no difference in the number of patients on any antihypertensive therapy (97% vs 99%). Interestingly, amputations were more common in the revascularization group, occurring in 42 (12%) of the 386 patients in the revascularization group vs 29 (7%) of the 395 patients in the medical group (P = .04).
Seventeen percent of patients randomized to intervention did not have the procedure done. An as-treated analysis of the 317 (83%) patients randomized to revascularization who did receive it showed no differences in outcomes.
There were no differences in outcomes among specific, predefined subgroups based on severity of stenosis at baseline, renal length, baseline estimated GFR, baseline serum creatinine, and rate of progression of renal dysfunction before randomization.7
Comments. ASTRAL contradicts previous nonrandomized studies that suggested that rapidly declining renal function (loss of 20% in 1 year) predicts response to intervention. Considering the large number of patients with unilateral disease in the study, it would be interesting to see what effect stenting had on patients with both severe disease and declining renal function.
ASTRAL has been criticized because it lacked a central laboratory to interpret the severity of stenosis, it did not use a standardized intervention technique (5% of patients underwent angioplasty without stents, although this did not affect outcomes7), and patients were enrolled only if the clinician involved in the case was uncertain of the appropriate management.
This last issue raises the concern for selection bias toward inclusion of more difficult cases that may not respond to intervention. But these shortcomings are not serious enough to negate the fact that preliminary results from the largest randomized controlled trial to date confirm conclusions of other randomized trials, ie, that intervention in renal artery stenosis yields no benefits over medical management in most patients.
Based on the results of STAR and ASTRAL, the practice of indiscriminately revascularizing stenosed renal arteries without strong evidence that the procedure will provide a clinical benefit is no longer tenable. The challenge is to identify those few patients who will respond, and to intervene only on them. Unfortunately, none of the subgroups from ASTRAL helped characterize this population.
CORAL: A large trial is ongoing
The Cardiovascular Outcomes in Renal Artherosclerotic Lesions (CORAL) trial,8 an ongoing multicenter randomized controlled trial in the United States, may be of additional help.
Unlike ASTRAL, CORAL is studying patients who have difficult-to-control hypertension (systolic blood pressure ≥ 155 mm Hg on two or more drugs).8 Chronic kidney disease is not an exclusion criterion unless the serum creatinine concentration is greater than 3.0 mg/dL.
CORAL is using a standardized medical protocol to control blood pressure. In addition, use of embolic protection devices during stenting is encouraged. Hopefully, the large size (a goal of 1,080 patients) and the inclusion of patients with more marked hypertension will address the utility of intervention in higher-risk populations with renal artery stenosis.
RECOMMENDED APPROACH TO INTERVENTION IN RENAL ARTERY STENOSIS
As we wait for CORAL to be completed, we have two modern-era randomized controlled trials that leave us with fewer indications for renal intervention. Table 2 lists commonly cited indications for intervention in renal artery stenosis and the evidence to support them. As most of these are based on retrospective data or have conflicting support in the literature, their utility remains in question. At this point we can safely recommend:
- Patients with preserved or even decreased but stable renal function will not likely have a benefit in renal function after intervention.
- Patients with resistant hypertension may benefit.
- The best evidence supporting intervention is for bilateral stenosis with flash pulmonary edema, but the evidence is from retrospective studies.
- Stenting in bilateral disease without another indication has no apparent benefit.
- Declining renal function is not a guarantee of success.
- It is unclear if patients with severe bilateral stenosis or severe stenosis to a solitary functioning kidney with declining renal function will benefit. Anecdotally, they do respond more often, but as with many other indications for intervention that have gone by the wayside, this may not bear out when studied properly.
As the utility of intervention narrows, the scope of practice for such interventions should narrow accordingly. Attention should now be focusing on clinical, rather than radiographic, indications for intervening on renal artery stenosis.
Therefore, the decision to intervene must not be made solely by the interventionalist. A multidisciplinary approach should be adopted that at the very least includes the input of a nephrologist well versed in renal artery stenosis. In this way, the clinical risks and benefits of renal intervention can be discussed with the patient by providers who are likely to be involved in their care should renal function or hypertension fail to improve afterward.
RISK OF ATHEROEMBOLISM
While renal stenting yields improved technical success in the treatment of renal artery stenosis, it carries with it an increasingly common risk to kidney function: atheroembolism as the stent crushes the plaque against the vessel wall. This may lead to obstruction of the renal microvasculature, increasing the risk of irreversible damage to renal function.
Atheroembolic kidney disease can manifest as progressive renal failure occurring over weeks to months, commonly misdiagnosed as permanent damage from contrast nephropathy.55
Embolic protection devices, inserted downstream of the lesion before stenting to catch any debris that may break loose, have been developed to help address this problem.
Holden et al 57 prospectively studied 63 patients with renal artery stenosis and deteriorating renal function (undefined) who underwent stenting with an embolic protection device. At 6 months after the intervention, renal function had either improved or stabilized in 97% of patients, suggesting that many of the deleterious effects of stenting on renal function are related to atheroembolism.
The Prospective Randomized Study Comparing Renal Artery Stenting With or Without Distal Protection (RESIST) trial, in which renal dysfunction was mild and the GFR was not declining (average estimated GFR 59.3 mL/min), found contrary results.57 In a two-by-two factorial study, patients were randomized to undergo stenting alone, stenting with the antiplatelet agent abciximab (ReoPro), stenting with an embolic protection device, or stenting with both abciximab and an embolic protection device. Interestingly, renal function declined in the first three groups, but remained stable in the group that received both abciximab and an embolic protection device.
ANTIPLATELET THERAPY AFTER RENAL STENTING: HOW LONG?
We have no data on the optimal duration of antiplatelet therapy after renal stenting, and guidelines from professional societies do not comment on it.58 As a result, practice patterns vary significantly among practitioners.
While in-stent restenosis rates are acceptably low after renal stenting, the risks and side effects of antiplatelet therapy often lead to arbitrary withdrawal of these drugs. The effect on stent patency is yet to be determined.
FUTURE DEVELOPMENTS
Results of STAR and ASTRAL confirm the growing suspicion that the surge seen in the last decade in renal artery stenting should be coming to an end. We await results either from CORAL or possibly a post hoc analysis of ASTRAL that might identify potential high-risk groups that will benefit from renal intervention. And as embolic protection devices become more agile and suitable to different renal lesions, there remains the possibility that, due to lower rates of unidentified atheroembolic kidney disease, CORAL may demonstrate improved renal outcomes after stenting. If not, the search for the best means to predict who should have renal intervention will continue.
We know through experience that stenting does provide great benefits for some patients with renal artery stenosis. Furthermore, the clinical problem is too intriguing, and too profitable, to die altogether.
- Choncol M, Linas S. Diagnosis and management of ischemic nephropathy. Clin J Am Soc Nephrol 2006; 1:172–181.
- Webster J, Marshall F, Abdalla M, et al Randomised comparison of percutaneous angioplasty vs continued medical therapy for hypertensive patients with atheromatous renal artery stenosis. Scottish and Newcastle Renal Artery Stenosis Collaborative Group. J Hum Hypertens 1998; 12:329–335.
- Plouin PF, Chatellier G, Darne B, Raynaud A. Blood pressure outcome of angioplasty in atherosclerotic renal artery stenosis: a randomized trial. Essai Multicentrique Medicaments vs Angioplastie (EMMA) Study Group. Hypertension 1998; 31:823–829.
- Textor SC. Revascularization in atherosclerotic renal artery disease. Kidney Int 1998; 53:799–811.
- Bax L, Woittiez AJ, Kouwenberg HJ, et al Stent placement in patients with atherosclerotic renal artery stenosis and impaired renal function: a randomized trial. Ann Intern Med 2009; 150:840–848.
- Mistry S, Ives N, Harding J, et al Angioplasty and STent for Renal Artery Lesions (ASTRAL trial): rationale, methods and results so far. J Hum Hypertens 2007; 21:511–515.
- Wheatley K, Ives N, Kalra P, Moss J. Revascularization versus medical therapy for renal-artery stenosis (ASTRAL). N Engl J Med 2009; 361:1953–1962.
- Cooper CJ, Murphy TP, Matsumoto A, et al Stent revascularization for the prevention of cardiovascular and renal events among patients with renal artery stenosis and systolic hypertension: rationale and design of the CORAL trial. Am Heart J 2006; 152:59–66.
- Galaria II, Surowiec SM, Rhodes JM, et al Percutaneous and open renal revascularizations have equivalent long-term functional outcomes. Ann Vasc Surg 2005; 19:218–228.
- Murphy TP, Soares G, Kim M. Increase in utilization of percutaneous renal artery interventions by Medicare beneficiaries 1996–2000. AJR Am J Roentgenol 2004; 183:561–568.
- Textor SC. Atherosclerotic renal artery stenosis: overtreated but underrated? J Am Soc Nephrol 2008; 19:656–659.
- Kalra PA, Guo H, Gilbertson DT, et al Atherosclerotic renovascular disease in the United States. Kidney Int 2010; 77:37–43.
- Hansen KJ, Edwards MS, Craven TE, et al Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002; 36:443–451.
- Cohen MG, Pascua JA, Garcia-Ben M, et al A simple prediction rule for significant renal artery stenosis in patients undergoing cardiac catheterization. Am Heart J 2005; 150:1204–1211.
- Buller CE, Nogareda JG, Ramanathan K, et al The profile of cardiac patients with renal artery stenosis. J Am Coll Cardiol 2004; 43:1606–1613.
- White CJ, Olin JW. Diagnosis and management of atherosclerotic renal artery stenosis: improving patient selection and outcomes. Nat Clin Pract Cardiovasc Med 2009; 6:176–190.
- Kalra PA, Guo H, Kausz AT, et al Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005; 69:293–301.
- Holley KE, Hunt JC, Brown AL, Kincaid OW, Sheps SG. Renal artery stenosis. A clinical-pathologic study in normotensive and hypertensive patients. Am J Med 1964; 37:14–22.
- de Mast Q, Beutler JJ. The prevalence of atherosclerotic renal artery stenosis in risk groups: a systemic literature review. J Hypertens 2009; 27:1333–1340.
- Kuczera P, Włoszczynska E, Adamczak M, Pencak P, Chudek J, Wiecek A. Frequency of renal artery stenosis and variants of renal vascularization in hypertensive patients: analysis of 1550 angiographies in one centre. J Hum Hypertens 2009; 23:396–401.
- Caps MT, Perissinotto C, Zierler RE, et al Prospective study of atherosclerotic disease progression in the renal artery. Circulation 1998; 98:2866–2872.
- Zierler RE, Bergelin RO, Davidson RC, Cantwell-Gab K, Polissar NL, Strandness DE. A prospective study of disease progression in patients with atherosclerotic renal artery stenosis. Am J Hypertens 1996; 9:1055–1061.
- Schreiber MJ, Pohl MA, Novick AC. The natural history of atherosclerotic and fibrous renal artery disease. Urol Clin North Am 1984; 11:383–392.
- Crowley JJ, Santos RM, Peter RH, et al Progression of renal artery stenosis in patients undergoing cardiac catheterization. Am Heart J 1998; 136:913–918.
- Textor SC. Renovascular hypertension update. Curr Hypertens Rep 2006; 8:521–527.
- Textor SC. Ischemic nephropathy: where are we now? J Am Soc Nephrol 2004; 15:1974–1982.
- Wright JR, Shurrab AE, Cheung C, et al A prospective study of the determinants of renal functional outcome and mortality in atherosclerotic renovascular disease. Am J Kidney Dis 2002; 39:1153–1161.
- Messina LM, Zelenock GB, Yao KA, Stanley JC. Renal revascularization for recurrent pulmonary edema in patients with poorly controlled hypertension and renal insufficiency: a distinct subgroup of patients with arteriosclerotic renal artery occlusive disease. J Vasc Surg 1992; 15:73–80.
- Bloch MJ, Trost DW, Pickering TG, Sos TA, August P. Prevention of recurrent pulmonary edema in patients with bilateral renovascular disease through renal artery stent placement. Am J Hypertens 1999; 12:1–7.
- Gray BH, Olin JW, Childs MB, Sullivan TM, Bacharach JM. Clinical benefit of renal artery angioplasty with stenting for the control of recurrent and refractory congestive heart failure. Vasc Med 2002; 7:275–279.
- Pickering TG, Herman L, Devereux RB, et al Recurrent pulmonary oedema in hypertension due to bilateral renal artery stenosis: treatment by angioplasty or surgical revascularisation. Lancet 1988; 2:551–552.
- Kennedy DJ, Colyer WR, Brewster PS, et al Renal insufficiency as a predictor of adverse events and mortality after renal artery stent placement. Am J Kidney Dis 2003; 14:926–935.
- Beutler JJ, Van Ampting JM, Van De Ven PJ, et al Long-term effects of arterial stenting on kidney function for patients with ostial atherosclerotic renal artery stenosis and renal insufficiency. J Am Soc Nephrol 2001; 12:1475–1481.
- Van de Ven PJ, Kaatee R, Beutler JJ, et al Arterial stenting and balloon angioplasty in ostial atherosclerotic renovascular disease: a randomized trial. Lancet 1999; 353:282–286.
- Isles C, Main J, O’Connell J, et al Survival associated with renovascular disease in Glasgow and Newcastle: a collaborative study. Scott Med J 1990; 35:70–73.
- Hunt JC, Sheps SG, Harrison EG, Strong CG, Bernatz PE. Renal and renovascular hypertension. A reasoned approach to diagnosis and management. Arch Intern Med 1974; 133:988–999.
- Textor SC. Atherosclerotic renal artery stenosis: how big is the problem, and what happens if nothing is done? J Hypertens Suppl 2005; 23:S5–S13.
- van Jaarsveld BC, Krijnen P, Pieterman H, et al The effect of balloon angioplasty on hypertension in atherosclerotic renal-artery stenosis. Dutch Renal Artery Stenosis Intervention Cooperative Study Group. N Engl J Med 2000; 342:1007–1014.
- Zeller T, Frank U, Müller C, et al Predictors of improved renal function after percutaneous stent-supported angioplasty of severe atherosclerotic ostial renal artery stenosis. Circulation 2003; 108;2244–2249.
- Chábová V, Schirger A, Stanson AW, McKusick MA, Textor SC. Outcomes of atherosclerotic renal artery stenosis managed without revascularization. Mayo Clin Proc 2000; 75:437–444.
- Davies MG, Saad WE, Peden EK, Mohiuddin IT, Naoum JJ, Lumsden AB. Implications of acute functional injury following percutaneous renal artery intervention. Ann Vasc Surg 2008; 22:783–789.
- Radermacher J, Chavin A, Bleck J, et al Use of Doppler ultrasonography to predict the outcome of therapy for renal-artery stenosis. N Eng J Med 2001; 344:410–417.
- García-Criado A, Gilabert R, Nicolau C, et al Value of Doppler sonography for predicting clinical outcome after renal artery revascularization in atherosclerotic renal artery stenosis. J Ultrasound Med 2005; 24:1641–1647.
- Zeller T, Müller C, Frank U, et al Stent angioplasty of severe atherosclerotic ostial renal artery stenosis in patients with diabetes mellitus and nephrosclerosis. Catheter Cardiovasc Interv 2003; 58:510–515.
- Harden PN, MacLeod MJ, Rodger RSC, et al Effect of renal-artery stenting on progression of renovascular renal failure. Lancet 1997; 349:1133–1136.
- Isles CG, Robertson S, Hill D. Management of renovascular disease: a review of renal artery stenting in ten studies. QJM 1999; 92:159–167.
- Muray S, Martın M, Amoedo ML, et al Rapid decline in renal function reflects reversibility and predicts the outcome after angioplasty in renal artery stenosis. Am J Kidney Dis 2002; 39:60–66.
- Textor SC, Glockner JF, Lerman LO, et al The use of magnetic resonance to evaluate tissue oxygenation in renal artery stenosis. J Am Soc Nephrol 2008; 19:780–788.
- Paraskevas KI, Perrea D, Briana DD, Liapis CD. Management of atherosclerotic renovascular disease: the effect of renal artery stenting on renal function and blood pressure. Int Urol Nephrol 2006; 38:683–691.
- Watson PS, Hadjipetrou P, Cox SV, Piemonte TC, Eisenhauer AC. Effect of renal artery stenting on renal function and size in patients with atherosclerotic renovascular disease. Circulation 2000; 102:1671–1677.
- Dean RH, Kieffer RW, Smith BM, et al Renovascular hypertension: anatomic and renal function changes during drug therapy. Arch Surg 1981; 116:1408–1415.
- Zhang Q, Shen W, Zhang R, Zhang J, Hu J, Zhang X. Effects of renal artery stenting on renal function and blood pressure in patients with atherosclerotic renovascular disease. Chin Med J (Engl) 2003; 116:1451–1454.
- Ramos F, Kotliar C, Alvarez D, et al Renal function and outcome of PTRA and stenting for atherosclerotic renal artery stenosis. Kidney Int 2003; 63:276–282.
- Rocha-Singh KJ, Ahuja RK, Sung CH, Rutherford J. Long-term renal function preservation after renal artery stenting in patients with progressive ischemic nephropathy. Catheter Cardiovasc Interv 2002; 57:135–141.
- Thadhani RI, Camargo CA, Xavier RJ, Fang LS, Bazari H. Atheroembolic renal failure after invasive procedures. Natural history based on 52 histologically proven cases. Medicine (Baltimore) 1995; 74:350–358.
- Holden A, Hill A, Jaff MR, Pilmore H. Renal artery stent revascularization with embolic protection in patients with ischemic nephropathy. Kidney Int 2006; 70:948–955.
- Cooper CJ, Haller ST, Colyer W, et al Embolic protection and platelet inhibition during renal artery stenting. Circulation 2008; 117:2752–2760.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al ACC/AHA 2005 Practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006; 113:e463–e654.
- Choncol M, Linas S. Diagnosis and management of ischemic nephropathy. Clin J Am Soc Nephrol 2006; 1:172–181.
- Webster J, Marshall F, Abdalla M, et al Randomised comparison of percutaneous angioplasty vs continued medical therapy for hypertensive patients with atheromatous renal artery stenosis. Scottish and Newcastle Renal Artery Stenosis Collaborative Group. J Hum Hypertens 1998; 12:329–335.
- Plouin PF, Chatellier G, Darne B, Raynaud A. Blood pressure outcome of angioplasty in atherosclerotic renal artery stenosis: a randomized trial. Essai Multicentrique Medicaments vs Angioplastie (EMMA) Study Group. Hypertension 1998; 31:823–829.
- Textor SC. Revascularization in atherosclerotic renal artery disease. Kidney Int 1998; 53:799–811.
- Bax L, Woittiez AJ, Kouwenberg HJ, et al Stent placement in patients with atherosclerotic renal artery stenosis and impaired renal function: a randomized trial. Ann Intern Med 2009; 150:840–848.
- Mistry S, Ives N, Harding J, et al Angioplasty and STent for Renal Artery Lesions (ASTRAL trial): rationale, methods and results so far. J Hum Hypertens 2007; 21:511–515.
- Wheatley K, Ives N, Kalra P, Moss J. Revascularization versus medical therapy for renal-artery stenosis (ASTRAL). N Engl J Med 2009; 361:1953–1962.
- Cooper CJ, Murphy TP, Matsumoto A, et al Stent revascularization for the prevention of cardiovascular and renal events among patients with renal artery stenosis and systolic hypertension: rationale and design of the CORAL trial. Am Heart J 2006; 152:59–66.
- Galaria II, Surowiec SM, Rhodes JM, et al Percutaneous and open renal revascularizations have equivalent long-term functional outcomes. Ann Vasc Surg 2005; 19:218–228.
- Murphy TP, Soares G, Kim M. Increase in utilization of percutaneous renal artery interventions by Medicare beneficiaries 1996–2000. AJR Am J Roentgenol 2004; 183:561–568.
- Textor SC. Atherosclerotic renal artery stenosis: overtreated but underrated? J Am Soc Nephrol 2008; 19:656–659.
- Kalra PA, Guo H, Gilbertson DT, et al Atherosclerotic renovascular disease in the United States. Kidney Int 2010; 77:37–43.
- Hansen KJ, Edwards MS, Craven TE, et al Prevalence of renovascular disease in the elderly: a population-based study. J Vasc Surg 2002; 36:443–451.
- Cohen MG, Pascua JA, Garcia-Ben M, et al A simple prediction rule for significant renal artery stenosis in patients undergoing cardiac catheterization. Am Heart J 2005; 150:1204–1211.
- Buller CE, Nogareda JG, Ramanathan K, et al The profile of cardiac patients with renal artery stenosis. J Am Coll Cardiol 2004; 43:1606–1613.
- White CJ, Olin JW. Diagnosis and management of atherosclerotic renal artery stenosis: improving patient selection and outcomes. Nat Clin Pract Cardiovasc Med 2009; 6:176–190.
- Kalra PA, Guo H, Kausz AT, et al Atherosclerotic renovascular disease in United States patients aged 67 years or older: risk factors, revascularization, and prognosis. Kidney Int 2005; 69:293–301.
- Holley KE, Hunt JC, Brown AL, Kincaid OW, Sheps SG. Renal artery stenosis. A clinical-pathologic study in normotensive and hypertensive patients. Am J Med 1964; 37:14–22.
- de Mast Q, Beutler JJ. The prevalence of atherosclerotic renal artery stenosis in risk groups: a systemic literature review. J Hypertens 2009; 27:1333–1340.
- Kuczera P, Włoszczynska E, Adamczak M, Pencak P, Chudek J, Wiecek A. Frequency of renal artery stenosis and variants of renal vascularization in hypertensive patients: analysis of 1550 angiographies in one centre. J Hum Hypertens 2009; 23:396–401.
- Caps MT, Perissinotto C, Zierler RE, et al Prospective study of atherosclerotic disease progression in the renal artery. Circulation 1998; 98:2866–2872.
- Zierler RE, Bergelin RO, Davidson RC, Cantwell-Gab K, Polissar NL, Strandness DE. A prospective study of disease progression in patients with atherosclerotic renal artery stenosis. Am J Hypertens 1996; 9:1055–1061.
- Schreiber MJ, Pohl MA, Novick AC. The natural history of atherosclerotic and fibrous renal artery disease. Urol Clin North Am 1984; 11:383–392.
- Crowley JJ, Santos RM, Peter RH, et al Progression of renal artery stenosis in patients undergoing cardiac catheterization. Am Heart J 1998; 136:913–918.
- Textor SC. Renovascular hypertension update. Curr Hypertens Rep 2006; 8:521–527.
- Textor SC. Ischemic nephropathy: where are we now? J Am Soc Nephrol 2004; 15:1974–1982.
- Wright JR, Shurrab AE, Cheung C, et al A prospective study of the determinants of renal functional outcome and mortality in atherosclerotic renovascular disease. Am J Kidney Dis 2002; 39:1153–1161.
- Messina LM, Zelenock GB, Yao KA, Stanley JC. Renal revascularization for recurrent pulmonary edema in patients with poorly controlled hypertension and renal insufficiency: a distinct subgroup of patients with arteriosclerotic renal artery occlusive disease. J Vasc Surg 1992; 15:73–80.
- Bloch MJ, Trost DW, Pickering TG, Sos TA, August P. Prevention of recurrent pulmonary edema in patients with bilateral renovascular disease through renal artery stent placement. Am J Hypertens 1999; 12:1–7.
- Gray BH, Olin JW, Childs MB, Sullivan TM, Bacharach JM. Clinical benefit of renal artery angioplasty with stenting for the control of recurrent and refractory congestive heart failure. Vasc Med 2002; 7:275–279.
- Pickering TG, Herman L, Devereux RB, et al Recurrent pulmonary oedema in hypertension due to bilateral renal artery stenosis: treatment by angioplasty or surgical revascularisation. Lancet 1988; 2:551–552.
- Kennedy DJ, Colyer WR, Brewster PS, et al Renal insufficiency as a predictor of adverse events and mortality after renal artery stent placement. Am J Kidney Dis 2003; 14:926–935.
- Beutler JJ, Van Ampting JM, Van De Ven PJ, et al Long-term effects of arterial stenting on kidney function for patients with ostial atherosclerotic renal artery stenosis and renal insufficiency. J Am Soc Nephrol 2001; 12:1475–1481.
- Van de Ven PJ, Kaatee R, Beutler JJ, et al Arterial stenting and balloon angioplasty in ostial atherosclerotic renovascular disease: a randomized trial. Lancet 1999; 353:282–286.
- Isles C, Main J, O’Connell J, et al Survival associated with renovascular disease in Glasgow and Newcastle: a collaborative study. Scott Med J 1990; 35:70–73.
- Hunt JC, Sheps SG, Harrison EG, Strong CG, Bernatz PE. Renal and renovascular hypertension. A reasoned approach to diagnosis and management. Arch Intern Med 1974; 133:988–999.
- Textor SC. Atherosclerotic renal artery stenosis: how big is the problem, and what happens if nothing is done? J Hypertens Suppl 2005; 23:S5–S13.
- van Jaarsveld BC, Krijnen P, Pieterman H, et al The effect of balloon angioplasty on hypertension in atherosclerotic renal-artery stenosis. Dutch Renal Artery Stenosis Intervention Cooperative Study Group. N Engl J Med 2000; 342:1007–1014.
- Zeller T, Frank U, Müller C, et al Predictors of improved renal function after percutaneous stent-supported angioplasty of severe atherosclerotic ostial renal artery stenosis. Circulation 2003; 108;2244–2249.
- Chábová V, Schirger A, Stanson AW, McKusick MA, Textor SC. Outcomes of atherosclerotic renal artery stenosis managed without revascularization. Mayo Clin Proc 2000; 75:437–444.
- Davies MG, Saad WE, Peden EK, Mohiuddin IT, Naoum JJ, Lumsden AB. Implications of acute functional injury following percutaneous renal artery intervention. Ann Vasc Surg 2008; 22:783–789.
- Radermacher J, Chavin A, Bleck J, et al Use of Doppler ultrasonography to predict the outcome of therapy for renal-artery stenosis. N Eng J Med 2001; 344:410–417.
- García-Criado A, Gilabert R, Nicolau C, et al Value of Doppler sonography for predicting clinical outcome after renal artery revascularization in atherosclerotic renal artery stenosis. J Ultrasound Med 2005; 24:1641–1647.
- Zeller T, Müller C, Frank U, et al Stent angioplasty of severe atherosclerotic ostial renal artery stenosis in patients with diabetes mellitus and nephrosclerosis. Catheter Cardiovasc Interv 2003; 58:510–515.
- Harden PN, MacLeod MJ, Rodger RSC, et al Effect of renal-artery stenting on progression of renovascular renal failure. Lancet 1997; 349:1133–1136.
- Isles CG, Robertson S, Hill D. Management of renovascular disease: a review of renal artery stenting in ten studies. QJM 1999; 92:159–167.
- Muray S, Martın M, Amoedo ML, et al Rapid decline in renal function reflects reversibility and predicts the outcome after angioplasty in renal artery stenosis. Am J Kidney Dis 2002; 39:60–66.
- Textor SC, Glockner JF, Lerman LO, et al The use of magnetic resonance to evaluate tissue oxygenation in renal artery stenosis. J Am Soc Nephrol 2008; 19:780–788.
- Paraskevas KI, Perrea D, Briana DD, Liapis CD. Management of atherosclerotic renovascular disease: the effect of renal artery stenting on renal function and blood pressure. Int Urol Nephrol 2006; 38:683–691.
- Watson PS, Hadjipetrou P, Cox SV, Piemonte TC, Eisenhauer AC. Effect of renal artery stenting on renal function and size in patients with atherosclerotic renovascular disease. Circulation 2000; 102:1671–1677.
- Dean RH, Kieffer RW, Smith BM, et al Renovascular hypertension: anatomic and renal function changes during drug therapy. Arch Surg 1981; 116:1408–1415.
- Zhang Q, Shen W, Zhang R, Zhang J, Hu J, Zhang X. Effects of renal artery stenting on renal function and blood pressure in patients with atherosclerotic renovascular disease. Chin Med J (Engl) 2003; 116:1451–1454.
- Ramos F, Kotliar C, Alvarez D, et al Renal function and outcome of PTRA and stenting for atherosclerotic renal artery stenosis. Kidney Int 2003; 63:276–282.
- Rocha-Singh KJ, Ahuja RK, Sung CH, Rutherford J. Long-term renal function preservation after renal artery stenting in patients with progressive ischemic nephropathy. Catheter Cardiovasc Interv 2002; 57:135–141.
- Thadhani RI, Camargo CA, Xavier RJ, Fang LS, Bazari H. Atheroembolic renal failure after invasive procedures. Natural history based on 52 histologically proven cases. Medicine (Baltimore) 1995; 74:350–358.
- Holden A, Hill A, Jaff MR, Pilmore H. Renal artery stent revascularization with embolic protection in patients with ischemic nephropathy. Kidney Int 2006; 70:948–955.
- Cooper CJ, Haller ST, Colyer W, et al Embolic protection and platelet inhibition during renal artery stenting. Circulation 2008; 117:2752–2760.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al ACC/AHA 2005 Practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006; 113:e463–e654.
KEY POINTS
- Two large randomized trials of intervention vs medical therapy showed negative results for intervention. A third trial is under way.
- Intervention is not recommended if renal function has remained stable over the past 6 to 12 months and if hypertension can be controlled medically.
- The best evidence supporting intervention is for bilateral stenosis with “flash” pulmonary edema, but the evidence is from retrospective studies.
- Stenosis by itself, even if bilateral, is not an indication for renal artery stenting.