User login
Inflammation as a link between brain injury and heart damage: The model of subarachnoid hemorrhage*
Subarachnoid hemorrhage (SAH) involves the rupture of an aneurysm in the deep part of the brain, around the circle of Willis, which disperses blood not within the parenchyma but around the brain. Despite this absence of parenchymal interaction, SAH is more potentially damaging than almost any other bleeding syndrome in the brain. Because of its association with heart disease, SAH has been at the nexus of investigation into heart-brain connections for a long time. As early as the 1940s and 1950s, a high incidence of cardiac problems, particularly electrocardiographic (ECG) abnormalities, was described in patients with SAH, especially in those with aneurysmal SAH.
SAH serves as a good model for studying heart-brain interactions because it is associated with both a high incidence of arrhythmia and a low prevalence of coronary heart disease. In a review of five major retrospective studies involving intervention for nontraumatic SAH, Lanzino and colleagues found that 91% of patients had evidence of atrial or ventricular arrhythmias on ECG.1 In a prospective study of 223 patients with SAH, Tung and colleagues found a low prevalence (5%) of preexisting cardiac disease.2 This latter finding suggests that the cardiac findings in patients with SAH are a unique phenomenon likely attributable to SAH itself, and this scarcity of confounding cardiac factors makes SAH an ideal model for heart-brain investigations. This review will discuss cardiac responses to cerebral injury in SAH and then look ahead to the use of a novel murine model of SAH to further examine these responses and explore their potential inflammatory underpinnings.
CARDIAC RESPONSES TO CEREBRAL INJURY IN PATIENTS WITH SUBARACHNOID HEMORRHAGE
Cardiac arrhythmias
Cardiac arrhythmias associated with SAH are common and well classified. Sakr and colleagues found rhythm abnormalities in 30.2% of 106 patients with SAH and an abnormal ECG; the most common rhythm abnormality was sinus bradycardia (16%), followed by sinus tachycardia (8.5%) and other arrhythmias (5.7%), which included ventricular premature contraction, ventricular bigeminy, and atrial fibrillation.3
Multifocal ventricular tachycardia (torsades de pointes) is associated with a high mortality rate and is a feared complication of SAH, but its importance has been called into question recently. Although Machado and colleagues found in a review of the literature that torsades de pointes occurred in 5 of 1,139 patients with SAH (0.4%), they were unable to rule out confounding factors (ie, hypokalemia and hypomagnesemia) as the cause of the arrhythmia.4 In a supportive finding, van den Bergh et al reported that QT intervals in patients with SAH are actually shorter when serum magnesium levels are lower (prolonged intervals are thought to indicate elevated risk for multifocal ventricular tachycardia).5 Although it is clear that patients with SAH frequently have a prolonged QT interval (discussed later), which is thought to be a risk factor for torsades de pointes, the electrolyte abnormalities seen in patients with SAH make it hard to definitively attribute the arrhythmia to the direct action of the brain.
Cardiac changes that resemble ischemia
Certain ECG changes seen in patients with SAH are referred to as ischemic changes because of their resemblance to ECG changes seen in acute coronary artery occlusion. In SAH, there is evidence that acute coronary artery occlusion is not present. The myocardial changes are assumed to be due to subendocardial ischemia. ECG abnormalities usually disappear within a few days or without resolution of the neurologic or cardiac condition. They are considered markers of the severity of SAH but not predictors for potentially serious cardiac complications or clinical outcomes.5
Repolarization abnormalities, also commonly seen in coronary artery ischemic disease, are common in SAH. Sakr et al found that 83% of patients with SAH developed repolarization abnormalities, with the most common being T-wave changes (39%) and the presence of U waves (26%).3 Deep, symmetric inverted T waves, usually without much ST-segment elevation or depression, are the typical abnormality. Left bundle branch block, which is sometimes considered a marker of acute, large-vessel ischemia, was present in only 2% of patients.3
Prolonged QT intervals were found in 34% of patients in the study by Sakr et al.3 The presence of this prolonged segment has become the most looked-for clinical tool for determining who might be at risk for cardiomyopathy. Although there is little evidence that the cardiomyopathy seen after SAH is associated closely with prolongation of the QT interval, it is a simple bedside sign that is readily available to all practitioners, given the practice of obtaining an ECG in almost all hospitalized patients at the time of admission.
In older patients with SAH, ECG changes occur with more severe events. In a retrospective study, Zaroff et al identified 439 patients with SAH, 58 of whom had ECG findings indicative of ischemia or myocardial infarction within 3 days of presentation and before surgery to correct an aneurysm.6 The most common ECG abnormality was T-wave inversions; the next most common abnormalities were ST depression, ST elevation, and Q waves of unknown duration. The most common pattern for ECG abnormalities suggests abnormalities in the anterior descending artery territory or in multiple vascular territories. Follow-up tracings demonstrating reversal of the abnormalities were available for 23 of the 58 patients (40%). There was no significant association between any specific ECG abnormality and mortality. Compared with patients with negative ECG findings, the patients with positive ECG findings were significantly older (mean age, 62 ± 15 years vs 53 ± 14 years), had a higher mean Hunt and Hess grade, and had higher all-cause mortality. Surprisingly, aneurysm location did not differ significantly between the two groups. These data suggest that coronary artery disease (which would be more common in the older population) may be a contributing factor to mortality.
CARDIOMYOPATHY
Regional or focal wall-motion abnormalities on echocardiogram have been observed in some patients with SAH, as have increased levels of creatine kinase, MB fraction (CK-MB). These findings often raise concern about ongoing cardiac ischemia from coronary artery disease and may cause treatment to be delayed. In our experience, patients who have undergone cardiac catheterization for this syndrome have been found not have coronary artery disease as the cause of their cardiac muscle damage.
There is a common misperception among trainees at our institution that patients who have coronary artery disease with neurologic causes do not have elevations in cardiac enzymes. This turns out not to be the case. Cardiac troponin I (cTnI) has been shown to be a more sensitive and specific marker for cardiac dysfunction in patients with SAH than is CK-MB.
In a study of 43 patients with SAH and no known coronary artery disease, Deibert et al found that 12 patients (28%) had elevated cTnI.7 Abnormal left ventricular function was apparent on echocardiogram in 7 of these 12 patients. cTnI proved to be 100% sensitive and 86% specific for detecting left ventricular dysfunction in patients with SAH in this study, whereas CK-MB was only 29% sensitive and 100% specific. Notably, all patients in whom left ventricular dysfunction developed returned to baseline function on follow-up studies.
Similarly, Parekh et al found that cTnI is elevated in 20% of patients with SAH and that these patients are more likely to manifest echocardiographic and clinical evidence of left ventricular dysfunction.8 Patients with more severe grades of SAH in this study were more likely to develop an elevated level of serum cTnI.
PATHOPHYSIOLOGY OF CARDIAC DYSFUNCTION IN SUBARACHNOID HEMORRHAGE
The pathophysiology of cardiac abnormalities in SAH is unsettled; one hypothesis that has support from human and experimental data proposes that sustained sympathetic stimulation of cardiomyocytes at the sympathetic nerve endings results in prolonged contraction and structural damage to the myocardium.5 Contraction band necrosis, a pathological pattern indicating that injury to the heart has occurred from muscles that have been energy-deprived from prolonged contraction, is a classic finding in autopsy specimens from patients with SAH. Transient low ejection fraction is the physiologic parameter that correlates with this pathologic finding.
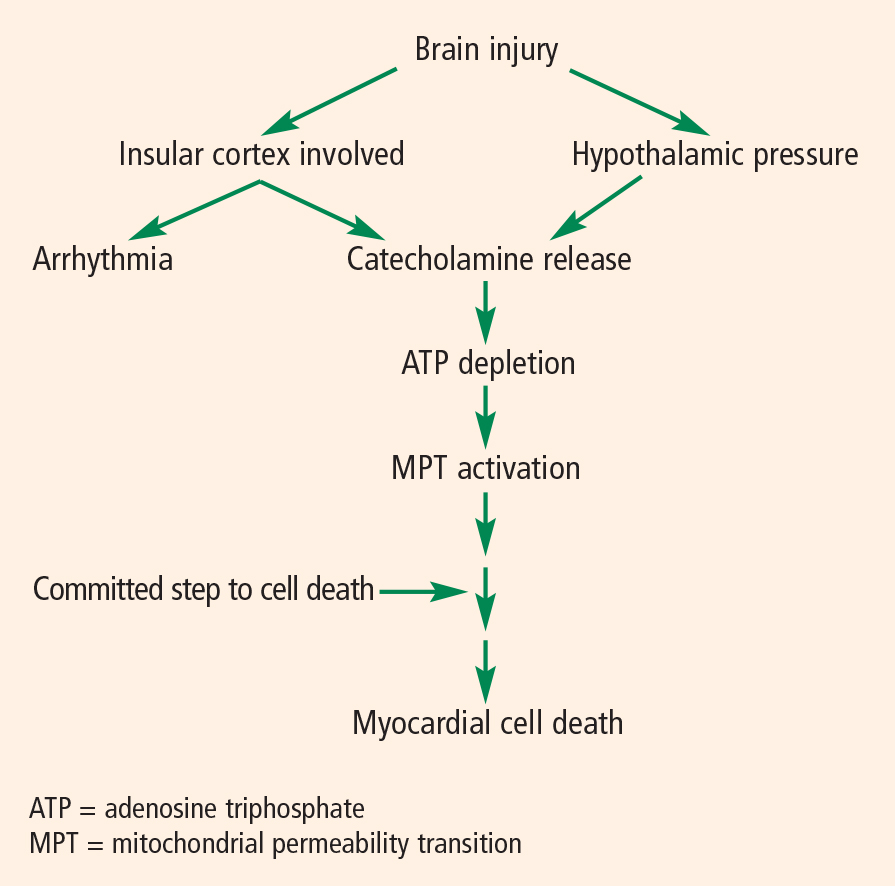
We recently presented an interesting finding that may suggest complementary mechanisms of cardiac dysfunction.10 Twenty-nine consecutive patients with SAH and no record of preexisting coronary artery disease were enrolled in a study of ECG abnormalities in SAH at Alexandria University Hospitals in Egypt. Each patient had ECGs during the preoperative period, during surgery, and during the first 3 days of postoperative treatment. We found that patients who had ECG abnormalities that fluctuated over the course of their early treatment had worse outcomes. This finding suggests that part of the mechanism of cardiac damage may occur later than the initial ictus.
The area that our laboratory has actively pursued is the interaction between the sympathetic nervous system, the parasympathetic nervous system, and inflammation in cardiac damage after SAH. There are reasons to believe that dysfunction of the parasympathetic system may be involved in the pathology of cardiac damage. The next section explains the underpinnings of why we believe this avenue of research needs to be explored.
ROLE OF VAGAL ACTIVITY AND INFLAMMATION
In the body, the sympathetic and parasympathetic systems work as the yin and yang in controlling many bodily functions. Rate and rhythm control of the heart is the prime example. Until relatively recently, the cardiac muscle was thought to be innervated predominantly by the sympathetic system, with the parasympathetic system largely innervating the conduction system. Fibers from the vagus nerve are now known to innervate the myocardium and therefore may play a role in the cardiac damage in SAH.
The role of the vagal system in inflammation is described elsewhere in this proceedings supplement. Briefly, there exists a recent body of research on the role of the vagal system in modulating the inflammatory system through acetylcholine receptors.11 The “neuorinflammatory reflex” (a term coined by Tracey11) is a vagally mediated phenomenon that may relate to parasympathetic nervous system activation (debate continues over whether this is a parasympathetic function or a function of the vagus nerve that is not autonomic) that suppresses inflammation.
Evidence of parasympathetic dysfunction in SAH is becoming more abundant. Kawahara et al measured heart rate variability in patients with acute SAH and determined that enhanced parasympathetic activity occurs acutely.9 This acute activation could potentially contribute to ECG abnormalities and cardiac injury. In addition, the parasympathetic response may also affect the inflammatory response. It has long been known that cardiomyopathy in patients with SAH and other brain traumas is accompanied by inflammation. It is unclear whether the neutrophil infiltration seen in this cardiac damage is due to the primary response from the brain (and therefore possibly contributory) or is in reaction to the cardiac damage.
Evidence from the transplant literature
Support for the role of inflammation in cardiac damage following SAH comes from the cardiac transplant literature. Data indicate that the cause of death in an organ donor has an impact on the organ recipient’s course of transplantation. Tsai et al compared outcomes among 251 transplant recipients who received hearts from donors who died of atraumatic intracranial bleeding (group 1; n = 80) or from donors who died of other causes (group 2; n = 171).12 They found that mortality among transplant recipients was higher in group 1 (14%) than in group 2 (5%).
Yamani et al performed cardiac biopsies 1 week after transplantation and then performed serial coronary intravascular ultrasonography over 1 year in 40 patients, half of whom received hearts from donors who died from intracerebral hemorrhage (ICH) and half from donors who died from trauma.13 At 1 week, heart biopsies from the ICH group showed greater expression of matrix metalloproteinases, enzymes that are responsible for matrix remodeling and associated with proinflammatory states, compared with biopsies from the trauma group. The injury in the ICH group translated to an increase in vasculopathy and myocardial fibrosis. At 1 year, hearts from donors who died of trauma had much less fibrosis and less progression of coronary vasculopathy (as measured by change in maximal intimal thickness on intravascular ultrasonography) than did hearts from donors who died from ICH, even after correction for differences in age.
Yamani et al also found that mRNA expression of angiotensin II type 1 receptor (AT1R), which is upregulated during acute inflammation, was elevated 4.7-fold in biopsies of transplanted hearts from the donors who died of ICH compared with the donors who died of trauma.14 There was likewise a 2.6-fold increase in AT1R mRNA expression in spleen lymphocytes from donors who died of ICH compared with donors who died from trauma, indicating that systemic activation of inflammation occurred before transplantation.14 AT1R mRNA expression has also been found to be seven times greater in the cerebrospinal fluid of patients with SAH than in a control population (unpublished data). The fact that upregulation of an inflammatory mediator in the heart of transplant recipients is associated with ICH suggests that there is a potential for the cerebral injury–induced inflammation seen in Tracey’s sepsis model11 to affect the heart in a setting other than sepsis.
A MODEL FOR SUBARACHNOID HEMORRHAGE
A murine model of SAH offers a number of advantages for studying the inflammatory underpinnings of cardiomyopathy. First, many of the immunological reagents needed to evaluate this problem are more easily available in mouse than in other species. Second, there are genetic manipulations of the inflammatory system that are more readily possible in mouse than in other species. Finally, at our institution, we have normative echocardiographic data that are better developed in the mouse than in other species.
A NEW MODEL FOR BRAIN-HEART INTERACTION
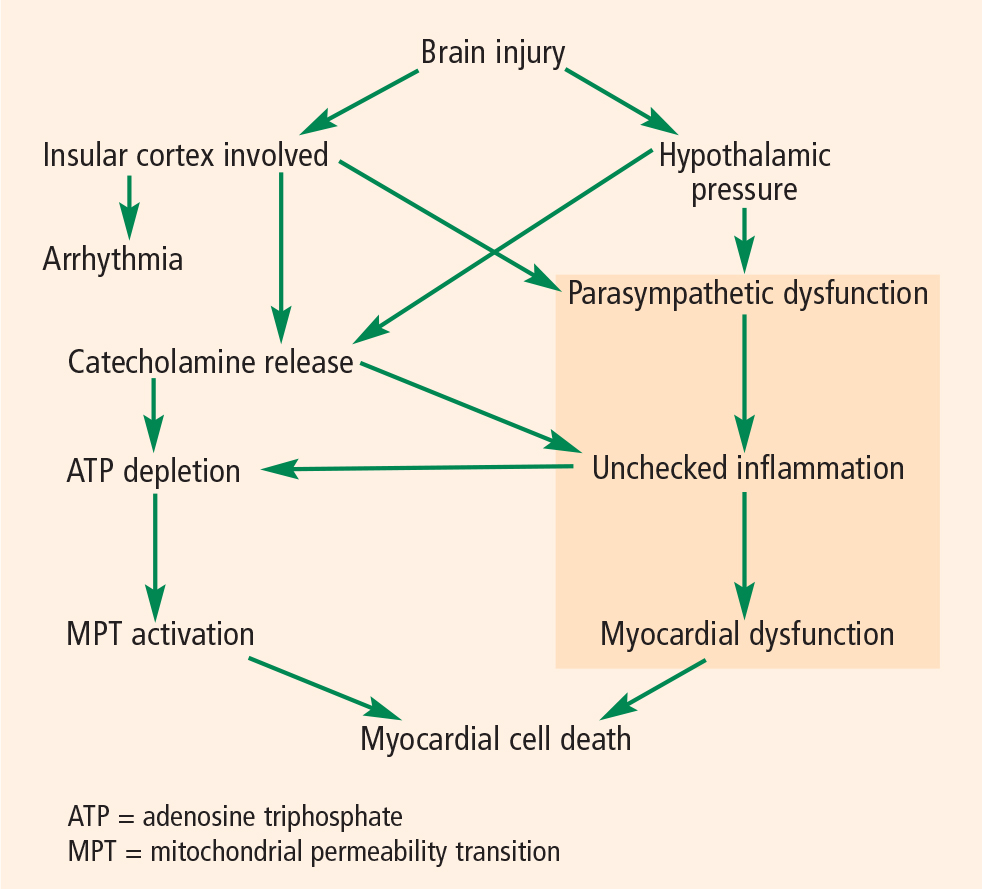
We hope that with better understanding of these two processes—ie, parasympathetic dysfunction and catecholamine release—we will be able to mitigate harm to the heart. If agents can be found that suppress sympathetic activation or heighten parasympathetic activation, it might be possible to improve outcomes in patients with SAH. This line of research will likely shape future efforts to further understand the pathophysiology of cardiac damage after brain injury and identify targets for clinical intervention.
- Lanzino G, Kongable GL, Kassell NF. Electrocardiographic abnormalities after nontraumatic subarachnoid hemorrhage. J Neurosurg Anesthesiol 1994; 6:156–162.
- Tung P, Kopelnik A, Banki N, et al. Predictors of neurocardiogenic injury after subarachnoid hemorrhage. Stroke 2004; 35:548–551.
- Sakr YL, Lim N, Amaral AC, et al. Relation of ECG changes to neurological outcome in patients with aneurysmal subarachnoid hemorrhage. Int J Cardiol 2004; 96:369–373.
- Machado C, Baga JJ, Kawasaki R, Reinoehl J, Steinman RT, Lehmann MH. Torsade de pointes as a complication of subarachnoid hemorrhage: a critical reappraisal. J Electrocardiol 1997; 30:31–37.
- van den Bergh WM, Algra A, Rinkel GJ. Electrocardiographic abnormalities and serum magnesium in patients with subarachnoid hemorrhage. Stroke 2004; 35:644–648.
- Zaroff JG, Rordorf GA, Newell JB, Ogilvy CS, Levinson JR. Cardiac outcome in patients with subarachnoid hemorrhage and electrocardiographic abnormalities. Neurosurgery 1999; 44:34–40.
- Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg 2003; 98:741–746.
- Parekh N, Venkatesh B, Cross D, et al. Cardiac troponin I predicts myocardial dysfunction in aneurysmal subarachnoid hemorrhage. J Am Coll Cardiol 2000; 36:1328–1335.
- Kawahara E, Ikeda S, Miyahara Y, Kohno S. Role of autonomic nervous dysfunction in electrocardiographic abnormalities and cardiac injury in patients with acute subarachnoid hemorrhage. Circ J 2003; 67:753–756.
- Elsharkawy HA, El Hadi SM, Tetzlaff JE, Provencio JJ. Dynamic changes in ECG predict poor outcome after aneurysmal subarachnoid hemorrhage (aSAH). Neurocrit Care (Supplement). In press.
- Tracey KJ. The inflammatory reflex. Nature 2002; 430:853–859.
- Tsai FC, Marelli D, Bresson J, et al; UCLA Heart Transplant Group. Use of hearts transplanted from donors with atraumatic intracranial bleeds. J Heart Lung Transplant 2002; 21:623–628.
- Yamani MH, Starling RC, Cook DJ, et al. Donor spontaneous intracerebral hemorrhage is associated with systemic activation of matrix metalloproteinase-2 and matrix metalloproteinase-9 and subsequent development of coronary vasculopathy in the heart transplant recipient. Circulation 2003; 108:1724–1728.
- Yamani MH, Cook DJ, Tuzcu EM, et al. Systemic up-regulation of angiontensin II type 1 receptor in cardiac donors with spontaneous intracerebral hemorrhage [published erratum appears in Am J Transplant 2004; 4:1928–1929]. Am J Transplant 2004; 4:1097–1102.
- Provencio JJ, Bleck TP. Cardiovascular disorders related to neurological and neurosurgical emergencies. In: Cruz J, ed. Neurological and Neurosurgical Emergencies. Philadelphia, PA: WB Saunders Co; 1998:39–50.
Subarachnoid hemorrhage (SAH) involves the rupture of an aneurysm in the deep part of the brain, around the circle of Willis, which disperses blood not within the parenchyma but around the brain. Despite this absence of parenchymal interaction, SAH is more potentially damaging than almost any other bleeding syndrome in the brain. Because of its association with heart disease, SAH has been at the nexus of investigation into heart-brain connections for a long time. As early as the 1940s and 1950s, a high incidence of cardiac problems, particularly electrocardiographic (ECG) abnormalities, was described in patients with SAH, especially in those with aneurysmal SAH.
SAH serves as a good model for studying heart-brain interactions because it is associated with both a high incidence of arrhythmia and a low prevalence of coronary heart disease. In a review of five major retrospective studies involving intervention for nontraumatic SAH, Lanzino and colleagues found that 91% of patients had evidence of atrial or ventricular arrhythmias on ECG.1 In a prospective study of 223 patients with SAH, Tung and colleagues found a low prevalence (5%) of preexisting cardiac disease.2 This latter finding suggests that the cardiac findings in patients with SAH are a unique phenomenon likely attributable to SAH itself, and this scarcity of confounding cardiac factors makes SAH an ideal model for heart-brain investigations. This review will discuss cardiac responses to cerebral injury in SAH and then look ahead to the use of a novel murine model of SAH to further examine these responses and explore their potential inflammatory underpinnings.
CARDIAC RESPONSES TO CEREBRAL INJURY IN PATIENTS WITH SUBARACHNOID HEMORRHAGE
Cardiac arrhythmias
Cardiac arrhythmias associated with SAH are common and well classified. Sakr and colleagues found rhythm abnormalities in 30.2% of 106 patients with SAH and an abnormal ECG; the most common rhythm abnormality was sinus bradycardia (16%), followed by sinus tachycardia (8.5%) and other arrhythmias (5.7%), which included ventricular premature contraction, ventricular bigeminy, and atrial fibrillation.3
Multifocal ventricular tachycardia (torsades de pointes) is associated with a high mortality rate and is a feared complication of SAH, but its importance has been called into question recently. Although Machado and colleagues found in a review of the literature that torsades de pointes occurred in 5 of 1,139 patients with SAH (0.4%), they were unable to rule out confounding factors (ie, hypokalemia and hypomagnesemia) as the cause of the arrhythmia.4 In a supportive finding, van den Bergh et al reported that QT intervals in patients with SAH are actually shorter when serum magnesium levels are lower (prolonged intervals are thought to indicate elevated risk for multifocal ventricular tachycardia).5 Although it is clear that patients with SAH frequently have a prolonged QT interval (discussed later), which is thought to be a risk factor for torsades de pointes, the electrolyte abnormalities seen in patients with SAH make it hard to definitively attribute the arrhythmia to the direct action of the brain.
Cardiac changes that resemble ischemia
Certain ECG changes seen in patients with SAH are referred to as ischemic changes because of their resemblance to ECG changes seen in acute coronary artery occlusion. In SAH, there is evidence that acute coronary artery occlusion is not present. The myocardial changes are assumed to be due to subendocardial ischemia. ECG abnormalities usually disappear within a few days or without resolution of the neurologic or cardiac condition. They are considered markers of the severity of SAH but not predictors for potentially serious cardiac complications or clinical outcomes.5
Repolarization abnormalities, also commonly seen in coronary artery ischemic disease, are common in SAH. Sakr et al found that 83% of patients with SAH developed repolarization abnormalities, with the most common being T-wave changes (39%) and the presence of U waves (26%).3 Deep, symmetric inverted T waves, usually without much ST-segment elevation or depression, are the typical abnormality. Left bundle branch block, which is sometimes considered a marker of acute, large-vessel ischemia, was present in only 2% of patients.3
Prolonged QT intervals were found in 34% of patients in the study by Sakr et al.3 The presence of this prolonged segment has become the most looked-for clinical tool for determining who might be at risk for cardiomyopathy. Although there is little evidence that the cardiomyopathy seen after SAH is associated closely with prolongation of the QT interval, it is a simple bedside sign that is readily available to all practitioners, given the practice of obtaining an ECG in almost all hospitalized patients at the time of admission.
In older patients with SAH, ECG changes occur with more severe events. In a retrospective study, Zaroff et al identified 439 patients with SAH, 58 of whom had ECG findings indicative of ischemia or myocardial infarction within 3 days of presentation and before surgery to correct an aneurysm.6 The most common ECG abnormality was T-wave inversions; the next most common abnormalities were ST depression, ST elevation, and Q waves of unknown duration. The most common pattern for ECG abnormalities suggests abnormalities in the anterior descending artery territory or in multiple vascular territories. Follow-up tracings demonstrating reversal of the abnormalities were available for 23 of the 58 patients (40%). There was no significant association between any specific ECG abnormality and mortality. Compared with patients with negative ECG findings, the patients with positive ECG findings were significantly older (mean age, 62 ± 15 years vs 53 ± 14 years), had a higher mean Hunt and Hess grade, and had higher all-cause mortality. Surprisingly, aneurysm location did not differ significantly between the two groups. These data suggest that coronary artery disease (which would be more common in the older population) may be a contributing factor to mortality.
CARDIOMYOPATHY
Regional or focal wall-motion abnormalities on echocardiogram have been observed in some patients with SAH, as have increased levels of creatine kinase, MB fraction (CK-MB). These findings often raise concern about ongoing cardiac ischemia from coronary artery disease and may cause treatment to be delayed. In our experience, patients who have undergone cardiac catheterization for this syndrome have been found not have coronary artery disease as the cause of their cardiac muscle damage.
There is a common misperception among trainees at our institution that patients who have coronary artery disease with neurologic causes do not have elevations in cardiac enzymes. This turns out not to be the case. Cardiac troponin I (cTnI) has been shown to be a more sensitive and specific marker for cardiac dysfunction in patients with SAH than is CK-MB.
In a study of 43 patients with SAH and no known coronary artery disease, Deibert et al found that 12 patients (28%) had elevated cTnI.7 Abnormal left ventricular function was apparent on echocardiogram in 7 of these 12 patients. cTnI proved to be 100% sensitive and 86% specific for detecting left ventricular dysfunction in patients with SAH in this study, whereas CK-MB was only 29% sensitive and 100% specific. Notably, all patients in whom left ventricular dysfunction developed returned to baseline function on follow-up studies.
Similarly, Parekh et al found that cTnI is elevated in 20% of patients with SAH and that these patients are more likely to manifest echocardiographic and clinical evidence of left ventricular dysfunction.8 Patients with more severe grades of SAH in this study were more likely to develop an elevated level of serum cTnI.
PATHOPHYSIOLOGY OF CARDIAC DYSFUNCTION IN SUBARACHNOID HEMORRHAGE
The pathophysiology of cardiac abnormalities in SAH is unsettled; one hypothesis that has support from human and experimental data proposes that sustained sympathetic stimulation of cardiomyocytes at the sympathetic nerve endings results in prolonged contraction and structural damage to the myocardium.5 Contraction band necrosis, a pathological pattern indicating that injury to the heart has occurred from muscles that have been energy-deprived from prolonged contraction, is a classic finding in autopsy specimens from patients with SAH. Transient low ejection fraction is the physiologic parameter that correlates with this pathologic finding.
We recently presented an interesting finding that may suggest complementary mechanisms of cardiac dysfunction.10 Twenty-nine consecutive patients with SAH and no record of preexisting coronary artery disease were enrolled in a study of ECG abnormalities in SAH at Alexandria University Hospitals in Egypt. Each patient had ECGs during the preoperative period, during surgery, and during the first 3 days of postoperative treatment. We found that patients who had ECG abnormalities that fluctuated over the course of their early treatment had worse outcomes. This finding suggests that part of the mechanism of cardiac damage may occur later than the initial ictus.
The area that our laboratory has actively pursued is the interaction between the sympathetic nervous system, the parasympathetic nervous system, and inflammation in cardiac damage after SAH. There are reasons to believe that dysfunction of the parasympathetic system may be involved in the pathology of cardiac damage. The next section explains the underpinnings of why we believe this avenue of research needs to be explored.
ROLE OF VAGAL ACTIVITY AND INFLAMMATION
In the body, the sympathetic and parasympathetic systems work as the yin and yang in controlling many bodily functions. Rate and rhythm control of the heart is the prime example. Until relatively recently, the cardiac muscle was thought to be innervated predominantly by the sympathetic system, with the parasympathetic system largely innervating the conduction system. Fibers from the vagus nerve are now known to innervate the myocardium and therefore may play a role in the cardiac damage in SAH.
The role of the vagal system in inflammation is described elsewhere in this proceedings supplement. Briefly, there exists a recent body of research on the role of the vagal system in modulating the inflammatory system through acetylcholine receptors.11 The “neuorinflammatory reflex” (a term coined by Tracey11) is a vagally mediated phenomenon that may relate to parasympathetic nervous system activation (debate continues over whether this is a parasympathetic function or a function of the vagus nerve that is not autonomic) that suppresses inflammation.
Evidence of parasympathetic dysfunction in SAH is becoming more abundant. Kawahara et al measured heart rate variability in patients with acute SAH and determined that enhanced parasympathetic activity occurs acutely.9 This acute activation could potentially contribute to ECG abnormalities and cardiac injury. In addition, the parasympathetic response may also affect the inflammatory response. It has long been known that cardiomyopathy in patients with SAH and other brain traumas is accompanied by inflammation. It is unclear whether the neutrophil infiltration seen in this cardiac damage is due to the primary response from the brain (and therefore possibly contributory) or is in reaction to the cardiac damage.
Evidence from the transplant literature
Support for the role of inflammation in cardiac damage following SAH comes from the cardiac transplant literature. Data indicate that the cause of death in an organ donor has an impact on the organ recipient’s course of transplantation. Tsai et al compared outcomes among 251 transplant recipients who received hearts from donors who died of atraumatic intracranial bleeding (group 1; n = 80) or from donors who died of other causes (group 2; n = 171).12 They found that mortality among transplant recipients was higher in group 1 (14%) than in group 2 (5%).
Yamani et al performed cardiac biopsies 1 week after transplantation and then performed serial coronary intravascular ultrasonography over 1 year in 40 patients, half of whom received hearts from donors who died from intracerebral hemorrhage (ICH) and half from donors who died from trauma.13 At 1 week, heart biopsies from the ICH group showed greater expression of matrix metalloproteinases, enzymes that are responsible for matrix remodeling and associated with proinflammatory states, compared with biopsies from the trauma group. The injury in the ICH group translated to an increase in vasculopathy and myocardial fibrosis. At 1 year, hearts from donors who died of trauma had much less fibrosis and less progression of coronary vasculopathy (as measured by change in maximal intimal thickness on intravascular ultrasonography) than did hearts from donors who died from ICH, even after correction for differences in age.
Yamani et al also found that mRNA expression of angiotensin II type 1 receptor (AT1R), which is upregulated during acute inflammation, was elevated 4.7-fold in biopsies of transplanted hearts from the donors who died of ICH compared with the donors who died of trauma.14 There was likewise a 2.6-fold increase in AT1R mRNA expression in spleen lymphocytes from donors who died of ICH compared with donors who died from trauma, indicating that systemic activation of inflammation occurred before transplantation.14 AT1R mRNA expression has also been found to be seven times greater in the cerebrospinal fluid of patients with SAH than in a control population (unpublished data). The fact that upregulation of an inflammatory mediator in the heart of transplant recipients is associated with ICH suggests that there is a potential for the cerebral injury–induced inflammation seen in Tracey’s sepsis model11 to affect the heart in a setting other than sepsis.
A MODEL FOR SUBARACHNOID HEMORRHAGE
A murine model of SAH offers a number of advantages for studying the inflammatory underpinnings of cardiomyopathy. First, many of the immunological reagents needed to evaluate this problem are more easily available in mouse than in other species. Second, there are genetic manipulations of the inflammatory system that are more readily possible in mouse than in other species. Finally, at our institution, we have normative echocardiographic data that are better developed in the mouse than in other species.
A NEW MODEL FOR BRAIN-HEART INTERACTION
We hope that with better understanding of these two processes—ie, parasympathetic dysfunction and catecholamine release—we will be able to mitigate harm to the heart. If agents can be found that suppress sympathetic activation or heighten parasympathetic activation, it might be possible to improve outcomes in patients with SAH. This line of research will likely shape future efforts to further understand the pathophysiology of cardiac damage after brain injury and identify targets for clinical intervention.
Subarachnoid hemorrhage (SAH) involves the rupture of an aneurysm in the deep part of the brain, around the circle of Willis, which disperses blood not within the parenchyma but around the brain. Despite this absence of parenchymal interaction, SAH is more potentially damaging than almost any other bleeding syndrome in the brain. Because of its association with heart disease, SAH has been at the nexus of investigation into heart-brain connections for a long time. As early as the 1940s and 1950s, a high incidence of cardiac problems, particularly electrocardiographic (ECG) abnormalities, was described in patients with SAH, especially in those with aneurysmal SAH.
SAH serves as a good model for studying heart-brain interactions because it is associated with both a high incidence of arrhythmia and a low prevalence of coronary heart disease. In a review of five major retrospective studies involving intervention for nontraumatic SAH, Lanzino and colleagues found that 91% of patients had evidence of atrial or ventricular arrhythmias on ECG.1 In a prospective study of 223 patients with SAH, Tung and colleagues found a low prevalence (5%) of preexisting cardiac disease.2 This latter finding suggests that the cardiac findings in patients with SAH are a unique phenomenon likely attributable to SAH itself, and this scarcity of confounding cardiac factors makes SAH an ideal model for heart-brain investigations. This review will discuss cardiac responses to cerebral injury in SAH and then look ahead to the use of a novel murine model of SAH to further examine these responses and explore their potential inflammatory underpinnings.
CARDIAC RESPONSES TO CEREBRAL INJURY IN PATIENTS WITH SUBARACHNOID HEMORRHAGE
Cardiac arrhythmias
Cardiac arrhythmias associated with SAH are common and well classified. Sakr and colleagues found rhythm abnormalities in 30.2% of 106 patients with SAH and an abnormal ECG; the most common rhythm abnormality was sinus bradycardia (16%), followed by sinus tachycardia (8.5%) and other arrhythmias (5.7%), which included ventricular premature contraction, ventricular bigeminy, and atrial fibrillation.3
Multifocal ventricular tachycardia (torsades de pointes) is associated with a high mortality rate and is a feared complication of SAH, but its importance has been called into question recently. Although Machado and colleagues found in a review of the literature that torsades de pointes occurred in 5 of 1,139 patients with SAH (0.4%), they were unable to rule out confounding factors (ie, hypokalemia and hypomagnesemia) as the cause of the arrhythmia.4 In a supportive finding, van den Bergh et al reported that QT intervals in patients with SAH are actually shorter when serum magnesium levels are lower (prolonged intervals are thought to indicate elevated risk for multifocal ventricular tachycardia).5 Although it is clear that patients with SAH frequently have a prolonged QT interval (discussed later), which is thought to be a risk factor for torsades de pointes, the electrolyte abnormalities seen in patients with SAH make it hard to definitively attribute the arrhythmia to the direct action of the brain.
Cardiac changes that resemble ischemia
Certain ECG changes seen in patients with SAH are referred to as ischemic changes because of their resemblance to ECG changes seen in acute coronary artery occlusion. In SAH, there is evidence that acute coronary artery occlusion is not present. The myocardial changes are assumed to be due to subendocardial ischemia. ECG abnormalities usually disappear within a few days or without resolution of the neurologic or cardiac condition. They are considered markers of the severity of SAH but not predictors for potentially serious cardiac complications or clinical outcomes.5
Repolarization abnormalities, also commonly seen in coronary artery ischemic disease, are common in SAH. Sakr et al found that 83% of patients with SAH developed repolarization abnormalities, with the most common being T-wave changes (39%) and the presence of U waves (26%).3 Deep, symmetric inverted T waves, usually without much ST-segment elevation or depression, are the typical abnormality. Left bundle branch block, which is sometimes considered a marker of acute, large-vessel ischemia, was present in only 2% of patients.3
Prolonged QT intervals were found in 34% of patients in the study by Sakr et al.3 The presence of this prolonged segment has become the most looked-for clinical tool for determining who might be at risk for cardiomyopathy. Although there is little evidence that the cardiomyopathy seen after SAH is associated closely with prolongation of the QT interval, it is a simple bedside sign that is readily available to all practitioners, given the practice of obtaining an ECG in almost all hospitalized patients at the time of admission.
In older patients with SAH, ECG changes occur with more severe events. In a retrospective study, Zaroff et al identified 439 patients with SAH, 58 of whom had ECG findings indicative of ischemia or myocardial infarction within 3 days of presentation and before surgery to correct an aneurysm.6 The most common ECG abnormality was T-wave inversions; the next most common abnormalities were ST depression, ST elevation, and Q waves of unknown duration. The most common pattern for ECG abnormalities suggests abnormalities in the anterior descending artery territory or in multiple vascular territories. Follow-up tracings demonstrating reversal of the abnormalities were available for 23 of the 58 patients (40%). There was no significant association between any specific ECG abnormality and mortality. Compared with patients with negative ECG findings, the patients with positive ECG findings were significantly older (mean age, 62 ± 15 years vs 53 ± 14 years), had a higher mean Hunt and Hess grade, and had higher all-cause mortality. Surprisingly, aneurysm location did not differ significantly between the two groups. These data suggest that coronary artery disease (which would be more common in the older population) may be a contributing factor to mortality.
CARDIOMYOPATHY
Regional or focal wall-motion abnormalities on echocardiogram have been observed in some patients with SAH, as have increased levels of creatine kinase, MB fraction (CK-MB). These findings often raise concern about ongoing cardiac ischemia from coronary artery disease and may cause treatment to be delayed. In our experience, patients who have undergone cardiac catheterization for this syndrome have been found not have coronary artery disease as the cause of their cardiac muscle damage.
There is a common misperception among trainees at our institution that patients who have coronary artery disease with neurologic causes do not have elevations in cardiac enzymes. This turns out not to be the case. Cardiac troponin I (cTnI) has been shown to be a more sensitive and specific marker for cardiac dysfunction in patients with SAH than is CK-MB.
In a study of 43 patients with SAH and no known coronary artery disease, Deibert et al found that 12 patients (28%) had elevated cTnI.7 Abnormal left ventricular function was apparent on echocardiogram in 7 of these 12 patients. cTnI proved to be 100% sensitive and 86% specific for detecting left ventricular dysfunction in patients with SAH in this study, whereas CK-MB was only 29% sensitive and 100% specific. Notably, all patients in whom left ventricular dysfunction developed returned to baseline function on follow-up studies.
Similarly, Parekh et al found that cTnI is elevated in 20% of patients with SAH and that these patients are more likely to manifest echocardiographic and clinical evidence of left ventricular dysfunction.8 Patients with more severe grades of SAH in this study were more likely to develop an elevated level of serum cTnI.
PATHOPHYSIOLOGY OF CARDIAC DYSFUNCTION IN SUBARACHNOID HEMORRHAGE
The pathophysiology of cardiac abnormalities in SAH is unsettled; one hypothesis that has support from human and experimental data proposes that sustained sympathetic stimulation of cardiomyocytes at the sympathetic nerve endings results in prolonged contraction and structural damage to the myocardium.5 Contraction band necrosis, a pathological pattern indicating that injury to the heart has occurred from muscles that have been energy-deprived from prolonged contraction, is a classic finding in autopsy specimens from patients with SAH. Transient low ejection fraction is the physiologic parameter that correlates with this pathologic finding.
We recently presented an interesting finding that may suggest complementary mechanisms of cardiac dysfunction.10 Twenty-nine consecutive patients with SAH and no record of preexisting coronary artery disease were enrolled in a study of ECG abnormalities in SAH at Alexandria University Hospitals in Egypt. Each patient had ECGs during the preoperative period, during surgery, and during the first 3 days of postoperative treatment. We found that patients who had ECG abnormalities that fluctuated over the course of their early treatment had worse outcomes. This finding suggests that part of the mechanism of cardiac damage may occur later than the initial ictus.
The area that our laboratory has actively pursued is the interaction between the sympathetic nervous system, the parasympathetic nervous system, and inflammation in cardiac damage after SAH. There are reasons to believe that dysfunction of the parasympathetic system may be involved in the pathology of cardiac damage. The next section explains the underpinnings of why we believe this avenue of research needs to be explored.
ROLE OF VAGAL ACTIVITY AND INFLAMMATION
In the body, the sympathetic and parasympathetic systems work as the yin and yang in controlling many bodily functions. Rate and rhythm control of the heart is the prime example. Until relatively recently, the cardiac muscle was thought to be innervated predominantly by the sympathetic system, with the parasympathetic system largely innervating the conduction system. Fibers from the vagus nerve are now known to innervate the myocardium and therefore may play a role in the cardiac damage in SAH.
The role of the vagal system in inflammation is described elsewhere in this proceedings supplement. Briefly, there exists a recent body of research on the role of the vagal system in modulating the inflammatory system through acetylcholine receptors.11 The “neuorinflammatory reflex” (a term coined by Tracey11) is a vagally mediated phenomenon that may relate to parasympathetic nervous system activation (debate continues over whether this is a parasympathetic function or a function of the vagus nerve that is not autonomic) that suppresses inflammation.
Evidence of parasympathetic dysfunction in SAH is becoming more abundant. Kawahara et al measured heart rate variability in patients with acute SAH and determined that enhanced parasympathetic activity occurs acutely.9 This acute activation could potentially contribute to ECG abnormalities and cardiac injury. In addition, the parasympathetic response may also affect the inflammatory response. It has long been known that cardiomyopathy in patients with SAH and other brain traumas is accompanied by inflammation. It is unclear whether the neutrophil infiltration seen in this cardiac damage is due to the primary response from the brain (and therefore possibly contributory) or is in reaction to the cardiac damage.
Evidence from the transplant literature
Support for the role of inflammation in cardiac damage following SAH comes from the cardiac transplant literature. Data indicate that the cause of death in an organ donor has an impact on the organ recipient’s course of transplantation. Tsai et al compared outcomes among 251 transplant recipients who received hearts from donors who died of atraumatic intracranial bleeding (group 1; n = 80) or from donors who died of other causes (group 2; n = 171).12 They found that mortality among transplant recipients was higher in group 1 (14%) than in group 2 (5%).
Yamani et al performed cardiac biopsies 1 week after transplantation and then performed serial coronary intravascular ultrasonography over 1 year in 40 patients, half of whom received hearts from donors who died from intracerebral hemorrhage (ICH) and half from donors who died from trauma.13 At 1 week, heart biopsies from the ICH group showed greater expression of matrix metalloproteinases, enzymes that are responsible for matrix remodeling and associated with proinflammatory states, compared with biopsies from the trauma group. The injury in the ICH group translated to an increase in vasculopathy and myocardial fibrosis. At 1 year, hearts from donors who died of trauma had much less fibrosis and less progression of coronary vasculopathy (as measured by change in maximal intimal thickness on intravascular ultrasonography) than did hearts from donors who died from ICH, even after correction for differences in age.
Yamani et al also found that mRNA expression of angiotensin II type 1 receptor (AT1R), which is upregulated during acute inflammation, was elevated 4.7-fold in biopsies of transplanted hearts from the donors who died of ICH compared with the donors who died of trauma.14 There was likewise a 2.6-fold increase in AT1R mRNA expression in spleen lymphocytes from donors who died of ICH compared with donors who died from trauma, indicating that systemic activation of inflammation occurred before transplantation.14 AT1R mRNA expression has also been found to be seven times greater in the cerebrospinal fluid of patients with SAH than in a control population (unpublished data). The fact that upregulation of an inflammatory mediator in the heart of transplant recipients is associated with ICH suggests that there is a potential for the cerebral injury–induced inflammation seen in Tracey’s sepsis model11 to affect the heart in a setting other than sepsis.
A MODEL FOR SUBARACHNOID HEMORRHAGE
A murine model of SAH offers a number of advantages for studying the inflammatory underpinnings of cardiomyopathy. First, many of the immunological reagents needed to evaluate this problem are more easily available in mouse than in other species. Second, there are genetic manipulations of the inflammatory system that are more readily possible in mouse than in other species. Finally, at our institution, we have normative echocardiographic data that are better developed in the mouse than in other species.
A NEW MODEL FOR BRAIN-HEART INTERACTION
We hope that with better understanding of these two processes—ie, parasympathetic dysfunction and catecholamine release—we will be able to mitigate harm to the heart. If agents can be found that suppress sympathetic activation or heighten parasympathetic activation, it might be possible to improve outcomes in patients with SAH. This line of research will likely shape future efforts to further understand the pathophysiology of cardiac damage after brain injury and identify targets for clinical intervention.
- Lanzino G, Kongable GL, Kassell NF. Electrocardiographic abnormalities after nontraumatic subarachnoid hemorrhage. J Neurosurg Anesthesiol 1994; 6:156–162.
- Tung P, Kopelnik A, Banki N, et al. Predictors of neurocardiogenic injury after subarachnoid hemorrhage. Stroke 2004; 35:548–551.
- Sakr YL, Lim N, Amaral AC, et al. Relation of ECG changes to neurological outcome in patients with aneurysmal subarachnoid hemorrhage. Int J Cardiol 2004; 96:369–373.
- Machado C, Baga JJ, Kawasaki R, Reinoehl J, Steinman RT, Lehmann MH. Torsade de pointes as a complication of subarachnoid hemorrhage: a critical reappraisal. J Electrocardiol 1997; 30:31–37.
- van den Bergh WM, Algra A, Rinkel GJ. Electrocardiographic abnormalities and serum magnesium in patients with subarachnoid hemorrhage. Stroke 2004; 35:644–648.
- Zaroff JG, Rordorf GA, Newell JB, Ogilvy CS, Levinson JR. Cardiac outcome in patients with subarachnoid hemorrhage and electrocardiographic abnormalities. Neurosurgery 1999; 44:34–40.
- Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg 2003; 98:741–746.
- Parekh N, Venkatesh B, Cross D, et al. Cardiac troponin I predicts myocardial dysfunction in aneurysmal subarachnoid hemorrhage. J Am Coll Cardiol 2000; 36:1328–1335.
- Kawahara E, Ikeda S, Miyahara Y, Kohno S. Role of autonomic nervous dysfunction in electrocardiographic abnormalities and cardiac injury in patients with acute subarachnoid hemorrhage. Circ J 2003; 67:753–756.
- Elsharkawy HA, El Hadi SM, Tetzlaff JE, Provencio JJ. Dynamic changes in ECG predict poor outcome after aneurysmal subarachnoid hemorrhage (aSAH). Neurocrit Care (Supplement). In press.
- Tracey KJ. The inflammatory reflex. Nature 2002; 430:853–859.
- Tsai FC, Marelli D, Bresson J, et al; UCLA Heart Transplant Group. Use of hearts transplanted from donors with atraumatic intracranial bleeds. J Heart Lung Transplant 2002; 21:623–628.
- Yamani MH, Starling RC, Cook DJ, et al. Donor spontaneous intracerebral hemorrhage is associated with systemic activation of matrix metalloproteinase-2 and matrix metalloproteinase-9 and subsequent development of coronary vasculopathy in the heart transplant recipient. Circulation 2003; 108:1724–1728.
- Yamani MH, Cook DJ, Tuzcu EM, et al. Systemic up-regulation of angiontensin II type 1 receptor in cardiac donors with spontaneous intracerebral hemorrhage [published erratum appears in Am J Transplant 2004; 4:1928–1929]. Am J Transplant 2004; 4:1097–1102.
- Provencio JJ, Bleck TP. Cardiovascular disorders related to neurological and neurosurgical emergencies. In: Cruz J, ed. Neurological and Neurosurgical Emergencies. Philadelphia, PA: WB Saunders Co; 1998:39–50.
- Lanzino G, Kongable GL, Kassell NF. Electrocardiographic abnormalities after nontraumatic subarachnoid hemorrhage. J Neurosurg Anesthesiol 1994; 6:156–162.
- Tung P, Kopelnik A, Banki N, et al. Predictors of neurocardiogenic injury after subarachnoid hemorrhage. Stroke 2004; 35:548–551.
- Sakr YL, Lim N, Amaral AC, et al. Relation of ECG changes to neurological outcome in patients with aneurysmal subarachnoid hemorrhage. Int J Cardiol 2004; 96:369–373.
- Machado C, Baga JJ, Kawasaki R, Reinoehl J, Steinman RT, Lehmann MH. Torsade de pointes as a complication of subarachnoid hemorrhage: a critical reappraisal. J Electrocardiol 1997; 30:31–37.
- van den Bergh WM, Algra A, Rinkel GJ. Electrocardiographic abnormalities and serum magnesium in patients with subarachnoid hemorrhage. Stroke 2004; 35:644–648.
- Zaroff JG, Rordorf GA, Newell JB, Ogilvy CS, Levinson JR. Cardiac outcome in patients with subarachnoid hemorrhage and electrocardiographic abnormalities. Neurosurgery 1999; 44:34–40.
- Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg 2003; 98:741–746.
- Parekh N, Venkatesh B, Cross D, et al. Cardiac troponin I predicts myocardial dysfunction in aneurysmal subarachnoid hemorrhage. J Am Coll Cardiol 2000; 36:1328–1335.
- Kawahara E, Ikeda S, Miyahara Y, Kohno S. Role of autonomic nervous dysfunction in electrocardiographic abnormalities and cardiac injury in patients with acute subarachnoid hemorrhage. Circ J 2003; 67:753–756.
- Elsharkawy HA, El Hadi SM, Tetzlaff JE, Provencio JJ. Dynamic changes in ECG predict poor outcome after aneurysmal subarachnoid hemorrhage (aSAH). Neurocrit Care (Supplement). In press.
- Tracey KJ. The inflammatory reflex. Nature 2002; 430:853–859.
- Tsai FC, Marelli D, Bresson J, et al; UCLA Heart Transplant Group. Use of hearts transplanted from donors with atraumatic intracranial bleeds. J Heart Lung Transplant 2002; 21:623–628.
- Yamani MH, Starling RC, Cook DJ, et al. Donor spontaneous intracerebral hemorrhage is associated with systemic activation of matrix metalloproteinase-2 and matrix metalloproteinase-9 and subsequent development of coronary vasculopathy in the heart transplant recipient. Circulation 2003; 108:1724–1728.
- Yamani MH, Cook DJ, Tuzcu EM, et al. Systemic up-regulation of angiontensin II type 1 receptor in cardiac donors with spontaneous intracerebral hemorrhage [published erratum appears in Am J Transplant 2004; 4:1928–1929]. Am J Transplant 2004; 4:1097–1102.
- Provencio JJ, Bleck TP. Cardiovascular disorders related to neurological and neurosurgical emergencies. In: Cruz J, ed. Neurological and Neurosurgical Emergencies. Philadelphia, PA: WB Saunders Co; 1998:39–50.