User login
Subacute Thyroiditis
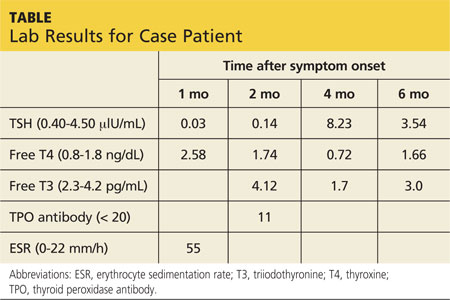
Jerry, a 48-year-old white man, is referred to endocrinology for abnormal results of thyroid tests performed four weeks ago (see table for values). Two months ago, Jerry developed an upper respiratory infection (URI) with fever, odynophagia, and anterior neck discomfort. His symptoms resolved after two weeks; however, he has since developed fatigue and nervousness.
The remaining review of systems is unremarkable. Medical history is negative. Jerry denies any factors that can affect thyroid function: He does not take thyroid medication, OTC thyroid supplements, amiodarone, lithium, or interferon-α, does not have high iodine intake, and has not undergone head/neck irradiation. There is no personal or family history of thyroid disease, organ-specific autoimmune disease (ie, vitiligo, myasthenia gravis, or Sjögren syndrome) or systemic autoimmune disease (rheumatoid arthritis, systemic lupus erythematosus, or progressive systemic sclerosis).
Vital signs are stable. On physical examination, his thyroid gland is firm, with slight enlargement of the left lobe and mild tenderness. There are no palpable nodules or cervical adenopathy. The remainder of the exam is unremarkable.
Lab studies (see table) reveal an elevated erythrocyte sedimentation rate (ESR) and suppressed TSH, with normal free thyroxine (T4) and free triiodothyronine (T3) levels. His thyroid peroxidase antibody (Anti-TPO) is negative. Radioactive iodine uptake (RAIU) reveals a low 24-hour uptake of 4% (normal, 5% to 30%).
Jerry is given the presumptive diagnosis of subacute thyroiditis (SAT). He is advised that the condition will progress through multiple phases—from the initial thyrotoxicosis to euthyroidism
to transient hypothyroid—before resolution and is educated on the symptoms and signs to watch for. Since he presented in a euthyroid phase, with only mild anterior neck tenderness, no treatment is indicated. He is instructed to follow up for thyroid function testing in four to six weeks and to call with any symptomatic changes.
Two months later, Jerry returns with complaints of ongoing fatigue, unintentional weight gain, and “mental fog.” Physical exam findings are unremarkable except for a small, firm thyroid gland without the tenderness elicited previously. Labwork reveals an elevated TSH with low free T4 and free T3. He is again counseled regarding the natural history of SAT and reassured that his symptoms will abate as his thyroid hormone levels normalize. He is advised to continue the plan of follow-up testing every four to six weeks.
Approximately eight weeks later, Jerry’s thyroid function studies indicate normal levels, and he is notified of the results. Jerry comments that his symptoms have completely resolved and he is back to feeling like his usual self. He is discharged to follow-up as needed.
What is subacute thyroiditis?
WHAT IS SUBACUTE THYROIDITIS?
Subacute thyroiditis is also known as de Quervain thyroiditis or granulomatous giant cell thyroiditis.1,2 The most common cause of thyroid pain, it is a self-limited inflammatory disorder in which a painful tender goiter is associated with malaise, fever, and transient thyroid dysfunction.2,3 As with other thyroid disorders, SAT occurs most frequently in women ages 40 to 50.2,3 Thought to be of viral origin, it usually occurs after a URI and commonly correlates with the peak incidence of viral infections (spring/fall).2,3
The disruptive process begins with inflammatory destruction of thyroid follicles.2 This causes leakage of stored colloid, which is broken down, releasing unregulated T4 and T3 into the circulation and resulting in a thyrotoxicosis that typically lasts six weeks.1,2,4 Thyroid cells are incapable of producing new thyroid hormone during this time, so as excess circulating hormone is utilized, T4 and T3 levels become normal, then deficient, and the patient transitions through a period of euthyroidism to transient hypothyroidism.1,2,4 As the disruption of thyroid parenchyma abates, recovery ensues. The follicles regenerate, colloid is repleted, and normal thyroid function is restored.1-4
SAT typically lasts four to six months, although painful thyromegaly may persist for one year after resolution of thyroid dysfunction.2 Throughout the course of SAT, thyroid test results can be confusing, and misdiagnosis of hyperthyroidism or hypothyroidism may occur unless each phase of SAT is recognized.
Phases of SAT >>
PRODROME
The precursor URI is followed in days or weeks by the clinical manifestations of SAT. These typically include myalgia, pharyngitis, low-grade fever, and fatigue.2
There may be pain of varying degrees in part or all of one or both lobes; the pain often migrates to the entire gland and may radiate to the angle of the jaw or the ear of the affected side(s). Moving the head, swallowing, or coughing aggravates the pain.2
The hallmark of SAT is a markedly elevated ESR (often > 100 mm/h).1-3 Leukocyte count is normal (50% of cases) or only slightly elevated (50%).2
THYROTOXIC PHASE
Fifty percent of patients have mild to moderate symptoms of hyperthyroidism, including nervousness, weight loss, heat intolerance, or palpitations; hoarseness or dysphagia may be present.2 Signs include tremors or tachycardia. The thyroid gland may reveal slight to moderate unilateral enlargement, usually firm in the involved area, and tenderness may be mild, moderate, or severe.2 Cervical lymphadenopathy is absent.2 Serum T4 and T3 levels are elevated, and TSH is suppressed.1-4
Thyroid antibodies (antithyroid peroxidase antibodies [Anti-TPO or TPOAb] or antithyroglobulin antibodies [Anti-TG or TgAb]) have been found in 42% to 62% of patients with SAT.2 These transitory immunologic markers develop several weeks after the onset and appear to be a physiologic response to the inflammatory insult to the gland.2 In most patients, the antibody titer gradually decreases, then disappears as the disease resolves.2-4
The 24-hour RAIU is low
(< 5%) in the toxic phase of SAT, and thyroid scan will reveal a patchy and irregular distribution of the tracer.2,3 The thyrotoxicosis during this early phase is caused by the inflammatory release of preformed thyroid hormones (not hyperfunctioning in the gland), resulting in a “low-uptake thyrotoxicosis.”2 This differentiates SAT from the elevated uptake seen in Graves disease (> 30% at 24 hours).2
TRANSIENT HYPOTHYROIDISM PHASE
As circulating T4 and T3 are utilized but follicular function remains temporarily impaired, levels decline, resulting in a period of euthyroidism followed by hypothyroidism. TSH levels, previously suppressed in the thyrotoxic phase, now become elevated. This transient hypothyroidism occurs in two-thirds of patients, and the presentation varies from subclinical to pronounced.2
RECOVERY PHASE
After several weeks or months, all thyroid function studies return to normal and complete recovery commonly ensues. SAT rarely recurs, most likely due to immunity to the precipitating virus.1,2,4
Management of SAT >>
MANAGEMENT
Thyroid function should be monitored by testing every two to four weeks, dependent on the severity of the patient’s symptoms and rate of progression.1 Often, no treatment is required.1,2
Symptomatic relief of mild thyroid pain can be achieved with NSAIDs or aspirin (2 to 3 g/d). Severe symptoms can be treated with short-term prednisone, which should be tapered and discontinued.1-3 Steroids suppress the inflammatory response, and the dramatic relief of thyroid pain within 24 hours can be diagnostic of SAT.2
During the thyrotoxic phase, β-blockers (propranolol) can alleviate adrenergic symptoms, with the dose tapered once the patient is euthyroid.1-3 Antithyroid medications that directly inhibit thyroid hormone synthesis (eg, methimazole or propylthiouracil) are ineffective due to the lack of T4 and T3 production in the follicular cells after the inflammatory response.2,3
During the transient hypothyroid phase, thyroid hormone replacement may be indicated if the TSH level is markedly elevated or the phase refractory. However, levothyroxine therapy should be low dose (< 100 μg) and not be considered lifelong.2,3
DIFFERENTIAL DIAGNOSIS
During the prodrome, SAT is often misdiagnosed as pharyngitis. Acute suppurative thyroiditis initially may mimic SAT, but the febrile and leukocytic responses are greater, and localized edema, erythema, and tenderness become more evident as the condition progresses.
Painless or silent thyroiditis is distinguished from SAT by the lack of pain or tenderness and a normal ESR in the presence of a similar pattern of thyroid dysfunction. Graves disease presents with symptoms similar to the thyrotoxic phase of SAT, but T3 is usually disproportionately elevated compared to T4, RAIU is elevated, and thyroid antibodies are prevalent.2
CONCLUSION
Primary care providers may encounter SAT at some point, and a level of clinical suspicion must be maintained. Referral to endocrinology may be warranted in some cases; however, textbook cases can often be followed in primary care. Patient education is the foundation of SAT care. Symptomatic treatments may be employed as needed. Fortunately, for most patients, this self-limited disease state rarely leads to complications.
REFERENCES
1. Cooper DS. The thyroid gland. In: Gardner D, Shobeck D (eds). Greenspan’s Basic and Clinical Endocrinology. 9th ed. China: McGraw-Hill; 2011:163-226.
2. Guimaraes VC. Subacute and Riedel’s thyroiditis. In: Jameson JL, De Groot LJ (eds). Endocrinology Adult and Pediatric. 6th ed. Philadelphia: Saunders; 2010:1595-1600.
3. Jameson JL. Disorders of the thyroid gland. In: Jameson JL (ed). Harrison’s Endocrinology. 2nd ed. China: McGraw-Hill; 2010: 62-98.
4. Smallridge RC. Thyroiditis. In: McDermott MT (ed). Endocrine Secrets. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:289-293.
Jerry, a 48-year-old white man, is referred to endocrinology for abnormal results of thyroid tests performed four weeks ago (see table for values). Two months ago, Jerry developed an upper respiratory infection (URI) with fever, odynophagia, and anterior neck discomfort. His symptoms resolved after two weeks; however, he has since developed fatigue and nervousness.
The remaining review of systems is unremarkable. Medical history is negative. Jerry denies any factors that can affect thyroid function: He does not take thyroid medication, OTC thyroid supplements, amiodarone, lithium, or interferon-α, does not have high iodine intake, and has not undergone head/neck irradiation. There is no personal or family history of thyroid disease, organ-specific autoimmune disease (ie, vitiligo, myasthenia gravis, or Sjögren syndrome) or systemic autoimmune disease (rheumatoid arthritis, systemic lupus erythematosus, or progressive systemic sclerosis).
Vital signs are stable. On physical examination, his thyroid gland is firm, with slight enlargement of the left lobe and mild tenderness. There are no palpable nodules or cervical adenopathy. The remainder of the exam is unremarkable.
Lab studies (see table) reveal an elevated erythrocyte sedimentation rate (ESR) and suppressed TSH, with normal free thyroxine (T4) and free triiodothyronine (T3) levels. His thyroid peroxidase antibody (Anti-TPO) is negative. Radioactive iodine uptake (RAIU) reveals a low 24-hour uptake of 4% (normal, 5% to 30%).
Jerry is given the presumptive diagnosis of subacute thyroiditis (SAT). He is advised that the condition will progress through multiple phases—from the initial thyrotoxicosis to euthyroidism
to transient hypothyroid—before resolution and is educated on the symptoms and signs to watch for. Since he presented in a euthyroid phase, with only mild anterior neck tenderness, no treatment is indicated. He is instructed to follow up for thyroid function testing in four to six weeks and to call with any symptomatic changes.
Two months later, Jerry returns with complaints of ongoing fatigue, unintentional weight gain, and “mental fog.” Physical exam findings are unremarkable except for a small, firm thyroid gland without the tenderness elicited previously. Labwork reveals an elevated TSH with low free T4 and free T3. He is again counseled regarding the natural history of SAT and reassured that his symptoms will abate as his thyroid hormone levels normalize. He is advised to continue the plan of follow-up testing every four to six weeks.
Approximately eight weeks later, Jerry’s thyroid function studies indicate normal levels, and he is notified of the results. Jerry comments that his symptoms have completely resolved and he is back to feeling like his usual self. He is discharged to follow-up as needed.
What is subacute thyroiditis?
WHAT IS SUBACUTE THYROIDITIS?
Subacute thyroiditis is also known as de Quervain thyroiditis or granulomatous giant cell thyroiditis.1,2 The most common cause of thyroid pain, it is a self-limited inflammatory disorder in which a painful tender goiter is associated with malaise, fever, and transient thyroid dysfunction.2,3 As with other thyroid disorders, SAT occurs most frequently in women ages 40 to 50.2,3 Thought to be of viral origin, it usually occurs after a URI and commonly correlates with the peak incidence of viral infections (spring/fall).2,3
The disruptive process begins with inflammatory destruction of thyroid follicles.2 This causes leakage of stored colloid, which is broken down, releasing unregulated T4 and T3 into the circulation and resulting in a thyrotoxicosis that typically lasts six weeks.1,2,4 Thyroid cells are incapable of producing new thyroid hormone during this time, so as excess circulating hormone is utilized, T4 and T3 levels become normal, then deficient, and the patient transitions through a period of euthyroidism to transient hypothyroidism.1,2,4 As the disruption of thyroid parenchyma abates, recovery ensues. The follicles regenerate, colloid is repleted, and normal thyroid function is restored.1-4
SAT typically lasts four to six months, although painful thyromegaly may persist for one year after resolution of thyroid dysfunction.2 Throughout the course of SAT, thyroid test results can be confusing, and misdiagnosis of hyperthyroidism or hypothyroidism may occur unless each phase of SAT is recognized.
Phases of SAT >>
PRODROME
The precursor URI is followed in days or weeks by the clinical manifestations of SAT. These typically include myalgia, pharyngitis, low-grade fever, and fatigue.2
There may be pain of varying degrees in part or all of one or both lobes; the pain often migrates to the entire gland and may radiate to the angle of the jaw or the ear of the affected side(s). Moving the head, swallowing, or coughing aggravates the pain.2
The hallmark of SAT is a markedly elevated ESR (often > 100 mm/h).1-3 Leukocyte count is normal (50% of cases) or only slightly elevated (50%).2
THYROTOXIC PHASE
Fifty percent of patients have mild to moderate symptoms of hyperthyroidism, including nervousness, weight loss, heat intolerance, or palpitations; hoarseness or dysphagia may be present.2 Signs include tremors or tachycardia. The thyroid gland may reveal slight to moderate unilateral enlargement, usually firm in the involved area, and tenderness may be mild, moderate, or severe.2 Cervical lymphadenopathy is absent.2 Serum T4 and T3 levels are elevated, and TSH is suppressed.1-4
Thyroid antibodies (antithyroid peroxidase antibodies [Anti-TPO or TPOAb] or antithyroglobulin antibodies [Anti-TG or TgAb]) have been found in 42% to 62% of patients with SAT.2 These transitory immunologic markers develop several weeks after the onset and appear to be a physiologic response to the inflammatory insult to the gland.2 In most patients, the antibody titer gradually decreases, then disappears as the disease resolves.2-4
The 24-hour RAIU is low
(< 5%) in the toxic phase of SAT, and thyroid scan will reveal a patchy and irregular distribution of the tracer.2,3 The thyrotoxicosis during this early phase is caused by the inflammatory release of preformed thyroid hormones (not hyperfunctioning in the gland), resulting in a “low-uptake thyrotoxicosis.”2 This differentiates SAT from the elevated uptake seen in Graves disease (> 30% at 24 hours).2
TRANSIENT HYPOTHYROIDISM PHASE
As circulating T4 and T3 are utilized but follicular function remains temporarily impaired, levels decline, resulting in a period of euthyroidism followed by hypothyroidism. TSH levels, previously suppressed in the thyrotoxic phase, now become elevated. This transient hypothyroidism occurs in two-thirds of patients, and the presentation varies from subclinical to pronounced.2
RECOVERY PHASE
After several weeks or months, all thyroid function studies return to normal and complete recovery commonly ensues. SAT rarely recurs, most likely due to immunity to the precipitating virus.1,2,4
Management of SAT >>
MANAGEMENT
Thyroid function should be monitored by testing every two to four weeks, dependent on the severity of the patient’s symptoms and rate of progression.1 Often, no treatment is required.1,2
Symptomatic relief of mild thyroid pain can be achieved with NSAIDs or aspirin (2 to 3 g/d). Severe symptoms can be treated with short-term prednisone, which should be tapered and discontinued.1-3 Steroids suppress the inflammatory response, and the dramatic relief of thyroid pain within 24 hours can be diagnostic of SAT.2
During the thyrotoxic phase, β-blockers (propranolol) can alleviate adrenergic symptoms, with the dose tapered once the patient is euthyroid.1-3 Antithyroid medications that directly inhibit thyroid hormone synthesis (eg, methimazole or propylthiouracil) are ineffective due to the lack of T4 and T3 production in the follicular cells after the inflammatory response.2,3
During the transient hypothyroid phase, thyroid hormone replacement may be indicated if the TSH level is markedly elevated or the phase refractory. However, levothyroxine therapy should be low dose (< 100 μg) and not be considered lifelong.2,3
DIFFERENTIAL DIAGNOSIS
During the prodrome, SAT is often misdiagnosed as pharyngitis. Acute suppurative thyroiditis initially may mimic SAT, but the febrile and leukocytic responses are greater, and localized edema, erythema, and tenderness become more evident as the condition progresses.
Painless or silent thyroiditis is distinguished from SAT by the lack of pain or tenderness and a normal ESR in the presence of a similar pattern of thyroid dysfunction. Graves disease presents with symptoms similar to the thyrotoxic phase of SAT, but T3 is usually disproportionately elevated compared to T4, RAIU is elevated, and thyroid antibodies are prevalent.2
CONCLUSION
Primary care providers may encounter SAT at some point, and a level of clinical suspicion must be maintained. Referral to endocrinology may be warranted in some cases; however, textbook cases can often be followed in primary care. Patient education is the foundation of SAT care. Symptomatic treatments may be employed as needed. Fortunately, for most patients, this self-limited disease state rarely leads to complications.
REFERENCES
1. Cooper DS. The thyroid gland. In: Gardner D, Shobeck D (eds). Greenspan’s Basic and Clinical Endocrinology. 9th ed. China: McGraw-Hill; 2011:163-226.
2. Guimaraes VC. Subacute and Riedel’s thyroiditis. In: Jameson JL, De Groot LJ (eds). Endocrinology Adult and Pediatric. 6th ed. Philadelphia: Saunders; 2010:1595-1600.
3. Jameson JL. Disorders of the thyroid gland. In: Jameson JL (ed). Harrison’s Endocrinology. 2nd ed. China: McGraw-Hill; 2010: 62-98.
4. Smallridge RC. Thyroiditis. In: McDermott MT (ed). Endocrine Secrets. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:289-293.
Jerry, a 48-year-old white man, is referred to endocrinology for abnormal results of thyroid tests performed four weeks ago (see table for values). Two months ago, Jerry developed an upper respiratory infection (URI) with fever, odynophagia, and anterior neck discomfort. His symptoms resolved after two weeks; however, he has since developed fatigue and nervousness.
The remaining review of systems is unremarkable. Medical history is negative. Jerry denies any factors that can affect thyroid function: He does not take thyroid medication, OTC thyroid supplements, amiodarone, lithium, or interferon-α, does not have high iodine intake, and has not undergone head/neck irradiation. There is no personal or family history of thyroid disease, organ-specific autoimmune disease (ie, vitiligo, myasthenia gravis, or Sjögren syndrome) or systemic autoimmune disease (rheumatoid arthritis, systemic lupus erythematosus, or progressive systemic sclerosis).
Vital signs are stable. On physical examination, his thyroid gland is firm, with slight enlargement of the left lobe and mild tenderness. There are no palpable nodules or cervical adenopathy. The remainder of the exam is unremarkable.
Lab studies (see table) reveal an elevated erythrocyte sedimentation rate (ESR) and suppressed TSH, with normal free thyroxine (T4) and free triiodothyronine (T3) levels. His thyroid peroxidase antibody (Anti-TPO) is negative. Radioactive iodine uptake (RAIU) reveals a low 24-hour uptake of 4% (normal, 5% to 30%).
Jerry is given the presumptive diagnosis of subacute thyroiditis (SAT). He is advised that the condition will progress through multiple phases—from the initial thyrotoxicosis to euthyroidism
to transient hypothyroid—before resolution and is educated on the symptoms and signs to watch for. Since he presented in a euthyroid phase, with only mild anterior neck tenderness, no treatment is indicated. He is instructed to follow up for thyroid function testing in four to six weeks and to call with any symptomatic changes.
Two months later, Jerry returns with complaints of ongoing fatigue, unintentional weight gain, and “mental fog.” Physical exam findings are unremarkable except for a small, firm thyroid gland without the tenderness elicited previously. Labwork reveals an elevated TSH with low free T4 and free T3. He is again counseled regarding the natural history of SAT and reassured that his symptoms will abate as his thyroid hormone levels normalize. He is advised to continue the plan of follow-up testing every four to six weeks.
Approximately eight weeks later, Jerry’s thyroid function studies indicate normal levels, and he is notified of the results. Jerry comments that his symptoms have completely resolved and he is back to feeling like his usual self. He is discharged to follow-up as needed.
What is subacute thyroiditis?
WHAT IS SUBACUTE THYROIDITIS?
Subacute thyroiditis is also known as de Quervain thyroiditis or granulomatous giant cell thyroiditis.1,2 The most common cause of thyroid pain, it is a self-limited inflammatory disorder in which a painful tender goiter is associated with malaise, fever, and transient thyroid dysfunction.2,3 As with other thyroid disorders, SAT occurs most frequently in women ages 40 to 50.2,3 Thought to be of viral origin, it usually occurs after a URI and commonly correlates with the peak incidence of viral infections (spring/fall).2,3
The disruptive process begins with inflammatory destruction of thyroid follicles.2 This causes leakage of stored colloid, which is broken down, releasing unregulated T4 and T3 into the circulation and resulting in a thyrotoxicosis that typically lasts six weeks.1,2,4 Thyroid cells are incapable of producing new thyroid hormone during this time, so as excess circulating hormone is utilized, T4 and T3 levels become normal, then deficient, and the patient transitions through a period of euthyroidism to transient hypothyroidism.1,2,4 As the disruption of thyroid parenchyma abates, recovery ensues. The follicles regenerate, colloid is repleted, and normal thyroid function is restored.1-4
SAT typically lasts four to six months, although painful thyromegaly may persist for one year after resolution of thyroid dysfunction.2 Throughout the course of SAT, thyroid test results can be confusing, and misdiagnosis of hyperthyroidism or hypothyroidism may occur unless each phase of SAT is recognized.
Phases of SAT >>
PRODROME
The precursor URI is followed in days or weeks by the clinical manifestations of SAT. These typically include myalgia, pharyngitis, low-grade fever, and fatigue.2
There may be pain of varying degrees in part or all of one or both lobes; the pain often migrates to the entire gland and may radiate to the angle of the jaw or the ear of the affected side(s). Moving the head, swallowing, or coughing aggravates the pain.2
The hallmark of SAT is a markedly elevated ESR (often > 100 mm/h).1-3 Leukocyte count is normal (50% of cases) or only slightly elevated (50%).2
THYROTOXIC PHASE
Fifty percent of patients have mild to moderate symptoms of hyperthyroidism, including nervousness, weight loss, heat intolerance, or palpitations; hoarseness or dysphagia may be present.2 Signs include tremors or tachycardia. The thyroid gland may reveal slight to moderate unilateral enlargement, usually firm in the involved area, and tenderness may be mild, moderate, or severe.2 Cervical lymphadenopathy is absent.2 Serum T4 and T3 levels are elevated, and TSH is suppressed.1-4
Thyroid antibodies (antithyroid peroxidase antibodies [Anti-TPO or TPOAb] or antithyroglobulin antibodies [Anti-TG or TgAb]) have been found in 42% to 62% of patients with SAT.2 These transitory immunologic markers develop several weeks after the onset and appear to be a physiologic response to the inflammatory insult to the gland.2 In most patients, the antibody titer gradually decreases, then disappears as the disease resolves.2-4
The 24-hour RAIU is low
(< 5%) in the toxic phase of SAT, and thyroid scan will reveal a patchy and irregular distribution of the tracer.2,3 The thyrotoxicosis during this early phase is caused by the inflammatory release of preformed thyroid hormones (not hyperfunctioning in the gland), resulting in a “low-uptake thyrotoxicosis.”2 This differentiates SAT from the elevated uptake seen in Graves disease (> 30% at 24 hours).2
TRANSIENT HYPOTHYROIDISM PHASE
As circulating T4 and T3 are utilized but follicular function remains temporarily impaired, levels decline, resulting in a period of euthyroidism followed by hypothyroidism. TSH levels, previously suppressed in the thyrotoxic phase, now become elevated. This transient hypothyroidism occurs in two-thirds of patients, and the presentation varies from subclinical to pronounced.2
RECOVERY PHASE
After several weeks or months, all thyroid function studies return to normal and complete recovery commonly ensues. SAT rarely recurs, most likely due to immunity to the precipitating virus.1,2,4
Management of SAT >>
MANAGEMENT
Thyroid function should be monitored by testing every two to four weeks, dependent on the severity of the patient’s symptoms and rate of progression.1 Often, no treatment is required.1,2
Symptomatic relief of mild thyroid pain can be achieved with NSAIDs or aspirin (2 to 3 g/d). Severe symptoms can be treated with short-term prednisone, which should be tapered and discontinued.1-3 Steroids suppress the inflammatory response, and the dramatic relief of thyroid pain within 24 hours can be diagnostic of SAT.2
During the thyrotoxic phase, β-blockers (propranolol) can alleviate adrenergic symptoms, with the dose tapered once the patient is euthyroid.1-3 Antithyroid medications that directly inhibit thyroid hormone synthesis (eg, methimazole or propylthiouracil) are ineffective due to the lack of T4 and T3 production in the follicular cells after the inflammatory response.2,3
During the transient hypothyroid phase, thyroid hormone replacement may be indicated if the TSH level is markedly elevated or the phase refractory. However, levothyroxine therapy should be low dose (< 100 μg) and not be considered lifelong.2,3
DIFFERENTIAL DIAGNOSIS
During the prodrome, SAT is often misdiagnosed as pharyngitis. Acute suppurative thyroiditis initially may mimic SAT, but the febrile and leukocytic responses are greater, and localized edema, erythema, and tenderness become more evident as the condition progresses.
Painless or silent thyroiditis is distinguished from SAT by the lack of pain or tenderness and a normal ESR in the presence of a similar pattern of thyroid dysfunction. Graves disease presents with symptoms similar to the thyrotoxic phase of SAT, but T3 is usually disproportionately elevated compared to T4, RAIU is elevated, and thyroid antibodies are prevalent.2
CONCLUSION
Primary care providers may encounter SAT at some point, and a level of clinical suspicion must be maintained. Referral to endocrinology may be warranted in some cases; however, textbook cases can often be followed in primary care. Patient education is the foundation of SAT care. Symptomatic treatments may be employed as needed. Fortunately, for most patients, this self-limited disease state rarely leads to complications.
REFERENCES
1. Cooper DS. The thyroid gland. In: Gardner D, Shobeck D (eds). Greenspan’s Basic and Clinical Endocrinology. 9th ed. China: McGraw-Hill; 2011:163-226.
2. Guimaraes VC. Subacute and Riedel’s thyroiditis. In: Jameson JL, De Groot LJ (eds). Endocrinology Adult and Pediatric. 6th ed. Philadelphia: Saunders; 2010:1595-1600.
3. Jameson JL. Disorders of the thyroid gland. In: Jameson JL (ed). Harrison’s Endocrinology. 2nd ed. China: McGraw-Hill; 2010: 62-98.
4. Smallridge RC. Thyroiditis. In: McDermott MT (ed). Endocrine Secrets. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:289-293.
New Standards of Care for Gestational Diabetes
Q: Why has there been a change in the standards of care for gestational diabetes mellitus?
With the obesity epidemic in this country, a greater number of women in their childbearing years are at risk for this continuum of metabolic syndrome, gestational diabetes mellitus (GDM), and type 2 diabetes mellitus (T2DM). GDM has long been defined as glucose intolerance with onset or first recognition during pregnancy.1 Compounding this definition are concerns that undiagnosed type 1 diabetes mellitus (T1DM) or T2DM may have been present at conception or that glucose intolerance may continue after pregnancy, developing into T2DM postpartum or prior to subsequent pregnancies.
In April 2011, the CDC reported that GDM occurs in 2% to 10% of pregnancies and is more likely in women who have a family history of diabetes, are obese, or are African-American, Hispanic/Latino American, or American Indian.2
Furthermore, in 2008, the Hyperglycemia and Adverse Pregnancy Outcomes (HAPO) study demonstrated a continuous association of adverse pregnancy outcomes (maternal, fetal, and neonatal) with increasing maternal glucose levels, even at glycemic levels previously considered normal.3 In response, the International Association of Diabetes and Pregnancy Study Groups (IADPSG) developed revised recommendations for diagnosing GDM; these were published in 2010.4 Professional organizations such as the American Diabetes Association (ADA) and the American Association of Clinical Endocrinologists (AACE) are now incorporating these revisions into their guidelines.5,6
Q: What causes GDM?
After 20 weeks’ gestation, there is an approximately 50% increase in insulin resistance in all women, most likely resulting from elevated levels of reproductive/placental hormones and possibly cytokines. As levels rise for the duration of the pregnancy, insulin resistance continually increases.
Women with normal pancreatic function are able to secrete the additional insulin required (as much as 200% to 250% by late pregnancy) to maintain euglycemia. Women predisposed to insulin resistance prior to pregnancy have already begun taxing their pancreatic beta-cells for increased production. As insulin resistance increases during the second and third trimesters, they are less likely to meet the additional demands for insulin, with impaired insulin secretion resulting in a greater risk for hyperglycemia.7
Q: What are the risks of maternal hyperglycemia for the child?
Hyperglycemia during the first trimester, either due to uncontrolled T1DM or undiagnosed T2DM, may cause birth defects, including neural tube and cardiac malformations; it can also contribute to early fetal loss.8,9 Maternal hyperglycemia during the second and third trimesters, as seen with the onset of GDM, is also associated with an increased risk for other complications, and HAPO reported that these complications increased with rising glucose levels previously considered normal.4
According to Pedersen’s hypothesis, maternal hyperglycemia transfers across the placenta, inducing fetal hyperglycemia, compensatory fetal hyperinsulinemia, and consequently increased adipose deposition of nutrients, resulting in macrosomia—the most common complication of GDM.10 Fetal hyperinsulinemia can siphon glucose from the mother, known as steal phenomenon, creating the appearance of maternal euglycemia.11 For this reason, ultrasound surveillance is used to monitor for accelerated fetal growth.
An estimated fetal weight of more than 4,500 g carries a high risk for shoulder dystocia with vaginal delivery, so elective cesarean section is usually recommended.12 Common complications in the infant include neonatal hypoglycemia secondary to fetal hyperinsulinemia, polycythemia, hyperbilirubinemia, and an increased need for neonatal intensive care.4 Furthermore, uncontrolled maternal GDM results in offspring at risk for childhood obesity and impaired glucose tolerance, giving rise to the theory of fetal imprinting and perpetuating a vicious cycle.13-16
In light of worldwide weight trends and with obesity considered the number one risk factor for insulin resistance, which is characterized by elevated triglycerides and free fatty acids, Catalano is revisiting Pedersen’s hypothesis, adding that the availability of maternal lipids for fetal lipogenesis is a possible contributing factor to fetal fat accumulation.17 This could explain why pregnancies complicated by obesity carry many of the same risks, or compound the maternal-fetal risks, that are associated with hyperglycemia in pregnancy.18-20
Q: What are the risks of GDM for the mother?
The immediate risks are an increased incidence of cesarean section, preeclampsia, and preterm delivery.3 Since GDM falls within the continuum for developing T2DM, pregnancy serves as a “stress test” in women who are genetically predisposed. Women who develop GDM have about a 50% risk for developing T2DM within five to 10 years.21
Q: How is GDM diagnosed?
As a result of the obesity and diabetes epidemics in this country, increasing numbers of women have undiagnosed T2DM with pregnancy.22 For this reason, the new guidelines recommend using standard diabetes screening for women with risk factors for T2DM at the first prenatal visit. Women who test positive are given a diagnosis of diabetes and should begin diabetes self-management education and therapeutic intervention, when indicated, without delay. AACE recommends that A1C be used as a screening test, due to potential interfering factors not related to glycemia, and that the diagnosis of diabetes be based on plasma glucose readings.6
Using HAPO data, the IADPSG proposed a new set of GDM diagnostic criteria in 2010, which has been adopted by ADA and AACE in 2011.3-6 Following the new standards, all women not previously diagnosed with diabetes should be screened at 24 to 28 weeks’ gestation using a 75-g, 2-h oral glucose tolerance test (OGTT) after an 8-h overnight fast. The diagnosis of GDM is made if there is one abnormal plasma glucose value: fasting glucose ≥ 92 mg/dL, 1-h glucose ≥ 180 mg/dL, or 2-h glucose ≥ 153 mg/dL.
Q: What are the goals for self-monitoring of blood glucose (SMBG) in pregnancy?
HAPO revealed that fasting and postprandial hyperglycemia all predicted risk for macrosomia, fetal hyperinsulinemia, and cesarean delivery.3 ADA and AACE concur with the recommendations from the Fifth International Workshop-Conference on Gestational Diabetes that SMBG goals for women with GDM are as follows: preprandial ≤ 95 mg/dL and either 1-h postprandial ≤ 140 mg/dL or 2-h postprandial ≤ 120 mg/dL.5,6,23
SMBG is recommended at least three times daily for pregnant women taking insulin, but ADA also recommends that monitoring be dictated by the needs and goals of the patient.5 For women with T1DM and T2DM who become pregnant, ADA and AACE concur with a recent consensus statement recommending avoidance of excessive hypoglycemia while aiming for glycemic goals of: preprandial, bedtime, and overnight glucose, 60 to 99 mg/dL, peak postprandial glucose, 100 to 129 mg/dL, and A1C 5,6,24
Q: What is the best therapy for women with GDM?
Although more women will be diagnosed with GDM using the 2011 guidelines, therapeutic lifestyle changes and medical nutrition therapy (diet) will be key elements in their management, as demonstrated in the Maternal-Fetal Medicine Units Network and the Australian Carbohydrate Intolerance Study in Pregnant Women trials.25,26 Women should not arbitrarily restrict calories, as ketosis can result from simply an overnight fast, and maternal ketones have been shown to impair the IQ of offspring.27,28
In many cases, diet alone, while preventing ketonuria, may control postprandial plasma glucose, but diet-control is less successful when fasting hyperglycemia develops. When SMBG does not meet the established goals, ADA, AACE, and the American College of Obstetricians and Gynecologists (ACOG) concur that insulin is the first-line therapy, either via multiple daily injections or continuous subcutaneous insulin infusion.5,6,12
Regular and neutral protamine hagedorn (NPH) insulin, both of which are classified as pregnancy category B, have been the mainstay therapy. Recently, rapid-acting insulin aspart has been approved for use in pregnancy, and lispro is considered a treatment option for patients with GDM by AACE and the 2008 Expert Review of Obstetrics and Gynecology on the Management of GDM.6,29 Likewise, 70/30 aspart mix and 75/25 lispro mix are now pregnancy category B. For basal insulin, detemir appears to be safe during pregnancy in early studies, while glargine, though used, has no conclusive reports on safety30,31; both remain pregnancy category C.
Recent studies have indicated that pregravid BMI should also be considered by clinicians who treat GDM. Results showed overweight women with GDM that is well controlled on diet alone had a 50% greater risk for delivering a macrosomic infant than did normal-weight patients with GDM, and this risk increased twofold for obese women. Of note, overweight and obese women with GDM that was well controlled on insulin had no increased risk for fetal macrosomia, compared to the reference group.32
The oral medications metformin (pregnancy category B) and glyburide, the most widely studied sulfonylurea (pregnancy category C), have been shown to be effective alternatives to insulin without adverse effects in some women.6 However, the studies have been limited.
Long-term effects on children exposed to these medications in utero are yet to be determined, and randomized, prospective studies using oral diabetic medications in pregnancy, with long-term follow-up of mothers and offspring, are needed. (Consequently, in our practice, we only use insulin therapy for GDM.)
Q: What is the follow-up for women diagnosed with GDM?
Due to the prevalence of T2DM in women with GDM, ADA, AACE, and ACOG concur with the recommendations from the Fifth International Workshop-Conference on Gestational Diabetes that a 75-g, 2-h OGTT be administered six to 12 weeks after delivery in women with GDM who do not have diabetes immediately postpartum.5,6,12,23 Thereafter, ADA recommends resuming routine screening every three years, with more frequent testing depending on initial results and risk status.5 Education regarding diet, weight loss, and exercise should be presented as lifestyle changes and adopted by the entire family, since the infant is also at risk for obesity and the metabolic syndrome.
REFERENCES
1. Metzger BE, Coustan DR. Summary and recommendations of the Fourth International Workshop Conference on Gestational Diabetes Mellitus. Diabetes Care. 1998;21(suppl 2):B161–B167.
2. CDC. National diabetes fact sheet, 2011. www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Accessed May 24, 2011.
3. Metzger BE, Lowe LP, Dyer AR, et al; HAPO Study Cooperative Research Group. Hyperglycemia and adverse pregnancy outcomes. N Engl J Med. 2008;358(19):1991-2002.
4. Metzger BE, Gabbe SG, Persson B, et al; International Association of Diabetes and Pregnancy Study Groups Consensus Panel. International Association of Diabetes and Pregnancy Study Groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care. 2010;33(3):676-682.
5. American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care. 2011;34(suppl 1):S11-S61.
6. Handelsman Y, Mechanick JI, Blonde L, et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for developing a diabetes mellitus comprehensive care plan. Endocr Pract. 2011;17(suppl 2):1-53.
7. Barbour LA, McCurdy CE, Hernandez TL, et al. Cellular mechanisms for insulin resistance in normal pregnancy and gestational diabetes. Diabetes Care. 2007;30(suppl 2):S112-S119.
8. Reece EA, Homko CJ. Why do diabetic women deliver malformed infants? Clin Obstet Gynecol. 2000;43(1):32-45.
9. Cundy T, Gamble G, Townend K, et al. Perinatal mortality in type 2 diabetes mellitus. Diabet Med. 2000;17(1):33-39.
10. Barbour LA. New concepts in insulin resistance of pregnancy and gestational diabetes: long-term implications for mother and offspring. J Obstet Gynaecol. 2003;23(5):545-549.
11. Weiss PA, Scholz HS, Haas J, Tamussino KF. Effect of fetal hyperinsulinism on oral glucose tolerance test results in patients with gestational diabetes mellitus. Am J Obstet Gynecol. 2001;184(3):470-475.
12. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin: clinical management guidelines for obstetrician-gynecologists. Obstet Gynecol. 2001;98(3):525-538.
13. Hillier TA, Pedula KL, Schmidt MM, et al. Childhood obesity and metabolic imprinting: the ongoing effects of maternal hyperglycemia. Diabetes Care. 2007;30(9):2287-2292.
14. Gillman MW, Rifas-Shiman S, Berkey CS, et al. Maternal gestational diabetes, birth weight, and adolescent obesity. Pediatrics. 2003;111(3):e221-e226.
15. Silverman BL, Rizzo TA, Cho NH, Metzger BE. Long-term effects of the intrauterine environment. Diabetes Care. 1998;21(suppl 2):B142- B149.
16. Pettitt DJ, Nelson RG, Saad MF, et al. Diabetes and obesity in the offspring of Pima Indian women with diabetes during pregnancy. Diabetes Care. 1993;16(1):310-314.
17. Catalano PM, Hauguel-de Mouzon S. Is it time to revisit the Pedersen hypothesis in the face of the obesity epidemic? Am J Obstet Gynecol. 2011 Feb 2; [Epub ahead of print].
18. Catalano PM. Obesity, insulin resistance, and pregnancy outcome. Reproduction. 2010;140(3):365-371.
19. Catalano PM, Presley L, Minium J, Hauguel-de Mouzon S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care. 2009;32(6):1076-1080.
20. Catalano PM. Obesity and pregnancy—the propagation of a viscous cycle? J Clin Endocrinol Metab. 2003;88(8):3505-3506.
21. Kim C, Newton KM, Knopp RH. Gestational diabetes and the incidence of type 2 diabetes: a systematic review. Diabetes Care. 2002;25(10):1862-1868.
22. Lawrence JM, Contreras R, Chen W, Sacks DA. Trends in the prevalence of preexisting diabetes and gestational diabetes mellitus among a racially/ethnically diverse population of pregnant women, 1999-2005. Diabetes Care. 2008;31(5):899-904.
23. Metzger BE, Buchanan TA, Coustan DR, et al. Summary and recommendations of the Fifth International Workshop-Conference on Gestational Diabetes Mellitus. Diabetes Care. 2007;30(suppl 2):S251-S260.
24. Kitzmiller JL, Block JM, Brown FM, et al. Managing preexisting diabetes for pregnancy: summary of evidence and consensus recommendations for care. Diabetes Care. 2008;31(5):1060-1079.
25. Landon MB, Spongy CY, Thorn E, et al; Eunice Kennedy Shriver National Institute of Child Health and Human Development Maternal-Fetal Medicine Units Network. A multicenter, randomized trial of treatment for mild gestational diabetes. N Engl J Med. 2009;361(14):1339-1348.
26. Crowther CA, Hiller JE, Moss JR, et al; Australian Carbohydrate Intolerance Study in Pregnant Women (ACHOIS) Trial Group. Effect of treatment of gestational diabetes mellitus on pregnancy outcomes. N Engl J Med. 2005;352(24):2477-2486.
27. Knopp RH, Magee MS, Raisys V, et al. Hypocaloric diets and ketogenesis in the management of obese gestational diabetic women. J Am Coll Nutr. 1991;10(6):649-667.
28. Rizzo T, Metzger BE, Burns WJ, Burns K. Correlations between antepartum maternal metabolism and child intelligence. N Engl J Med. 1991; 325(13):911-916.
29. Menato G, Bo S, Signorile A, et al. Current management of gestational diabetes mellitus. Expert Rev of Obstet Gynecol. 2008;3(1):73-91.
30. Sciacca L, Marotta V, Insalaco F, et al. Use of insulin detemir during pregnancy. Nutr Metab Cardiovasc Dis. 2010;20(4):e15-e16.
31. Lapolla A, Di Cianni G, Bruttomesso D, et al. Use of insulin detemir in pregnancy: a report on 10 type 1 diabetic women. Diabet Med. 2009; 26(11):1181-1182.
32. Langer O, Yogev Y, Xenakis EM, Brustman L. Overweight and obese gestational diabetes: impact on pregnancy outcome. Am J Obstet Gynecol. 2005;192(6):1768-1776.
Q: Why has there been a change in the standards of care for gestational diabetes mellitus?
With the obesity epidemic in this country, a greater number of women in their childbearing years are at risk for this continuum of metabolic syndrome, gestational diabetes mellitus (GDM), and type 2 diabetes mellitus (T2DM). GDM has long been defined as glucose intolerance with onset or first recognition during pregnancy.1 Compounding this definition are concerns that undiagnosed type 1 diabetes mellitus (T1DM) or T2DM may have been present at conception or that glucose intolerance may continue after pregnancy, developing into T2DM postpartum or prior to subsequent pregnancies.
In April 2011, the CDC reported that GDM occurs in 2% to 10% of pregnancies and is more likely in women who have a family history of diabetes, are obese, or are African-American, Hispanic/Latino American, or American Indian.2
Furthermore, in 2008, the Hyperglycemia and Adverse Pregnancy Outcomes (HAPO) study demonstrated a continuous association of adverse pregnancy outcomes (maternal, fetal, and neonatal) with increasing maternal glucose levels, even at glycemic levels previously considered normal.3 In response, the International Association of Diabetes and Pregnancy Study Groups (IADPSG) developed revised recommendations for diagnosing GDM; these were published in 2010.4 Professional organizations such as the American Diabetes Association (ADA) and the American Association of Clinical Endocrinologists (AACE) are now incorporating these revisions into their guidelines.5,6
Q: What causes GDM?
After 20 weeks’ gestation, there is an approximately 50% increase in insulin resistance in all women, most likely resulting from elevated levels of reproductive/placental hormones and possibly cytokines. As levels rise for the duration of the pregnancy, insulin resistance continually increases.
Women with normal pancreatic function are able to secrete the additional insulin required (as much as 200% to 250% by late pregnancy) to maintain euglycemia. Women predisposed to insulin resistance prior to pregnancy have already begun taxing their pancreatic beta-cells for increased production. As insulin resistance increases during the second and third trimesters, they are less likely to meet the additional demands for insulin, with impaired insulin secretion resulting in a greater risk for hyperglycemia.7
Q: What are the risks of maternal hyperglycemia for the child?
Hyperglycemia during the first trimester, either due to uncontrolled T1DM or undiagnosed T2DM, may cause birth defects, including neural tube and cardiac malformations; it can also contribute to early fetal loss.8,9 Maternal hyperglycemia during the second and third trimesters, as seen with the onset of GDM, is also associated with an increased risk for other complications, and HAPO reported that these complications increased with rising glucose levels previously considered normal.4
According to Pedersen’s hypothesis, maternal hyperglycemia transfers across the placenta, inducing fetal hyperglycemia, compensatory fetal hyperinsulinemia, and consequently increased adipose deposition of nutrients, resulting in macrosomia—the most common complication of GDM.10 Fetal hyperinsulinemia can siphon glucose from the mother, known as steal phenomenon, creating the appearance of maternal euglycemia.11 For this reason, ultrasound surveillance is used to monitor for accelerated fetal growth.
An estimated fetal weight of more than 4,500 g carries a high risk for shoulder dystocia with vaginal delivery, so elective cesarean section is usually recommended.12 Common complications in the infant include neonatal hypoglycemia secondary to fetal hyperinsulinemia, polycythemia, hyperbilirubinemia, and an increased need for neonatal intensive care.4 Furthermore, uncontrolled maternal GDM results in offspring at risk for childhood obesity and impaired glucose tolerance, giving rise to the theory of fetal imprinting and perpetuating a vicious cycle.13-16
In light of worldwide weight trends and with obesity considered the number one risk factor for insulin resistance, which is characterized by elevated triglycerides and free fatty acids, Catalano is revisiting Pedersen’s hypothesis, adding that the availability of maternal lipids for fetal lipogenesis is a possible contributing factor to fetal fat accumulation.17 This could explain why pregnancies complicated by obesity carry many of the same risks, or compound the maternal-fetal risks, that are associated with hyperglycemia in pregnancy.18-20
Q: What are the risks of GDM for the mother?
The immediate risks are an increased incidence of cesarean section, preeclampsia, and preterm delivery.3 Since GDM falls within the continuum for developing T2DM, pregnancy serves as a “stress test” in women who are genetically predisposed. Women who develop GDM have about a 50% risk for developing T2DM within five to 10 years.21
Q: How is GDM diagnosed?
As a result of the obesity and diabetes epidemics in this country, increasing numbers of women have undiagnosed T2DM with pregnancy.22 For this reason, the new guidelines recommend using standard diabetes screening for women with risk factors for T2DM at the first prenatal visit. Women who test positive are given a diagnosis of diabetes and should begin diabetes self-management education and therapeutic intervention, when indicated, without delay. AACE recommends that A1C be used as a screening test, due to potential interfering factors not related to glycemia, and that the diagnosis of diabetes be based on plasma glucose readings.6
Using HAPO data, the IADPSG proposed a new set of GDM diagnostic criteria in 2010, which has been adopted by ADA and AACE in 2011.3-6 Following the new standards, all women not previously diagnosed with diabetes should be screened at 24 to 28 weeks’ gestation using a 75-g, 2-h oral glucose tolerance test (OGTT) after an 8-h overnight fast. The diagnosis of GDM is made if there is one abnormal plasma glucose value: fasting glucose ≥ 92 mg/dL, 1-h glucose ≥ 180 mg/dL, or 2-h glucose ≥ 153 mg/dL.
Q: What are the goals for self-monitoring of blood glucose (SMBG) in pregnancy?
HAPO revealed that fasting and postprandial hyperglycemia all predicted risk for macrosomia, fetal hyperinsulinemia, and cesarean delivery.3 ADA and AACE concur with the recommendations from the Fifth International Workshop-Conference on Gestational Diabetes that SMBG goals for women with GDM are as follows: preprandial ≤ 95 mg/dL and either 1-h postprandial ≤ 140 mg/dL or 2-h postprandial ≤ 120 mg/dL.5,6,23
SMBG is recommended at least three times daily for pregnant women taking insulin, but ADA also recommends that monitoring be dictated by the needs and goals of the patient.5 For women with T1DM and T2DM who become pregnant, ADA and AACE concur with a recent consensus statement recommending avoidance of excessive hypoglycemia while aiming for glycemic goals of: preprandial, bedtime, and overnight glucose, 60 to 99 mg/dL, peak postprandial glucose, 100 to 129 mg/dL, and A1C 5,6,24
Q: What is the best therapy for women with GDM?
Although more women will be diagnosed with GDM using the 2011 guidelines, therapeutic lifestyle changes and medical nutrition therapy (diet) will be key elements in their management, as demonstrated in the Maternal-Fetal Medicine Units Network and the Australian Carbohydrate Intolerance Study in Pregnant Women trials.25,26 Women should not arbitrarily restrict calories, as ketosis can result from simply an overnight fast, and maternal ketones have been shown to impair the IQ of offspring.27,28
In many cases, diet alone, while preventing ketonuria, may control postprandial plasma glucose, but diet-control is less successful when fasting hyperglycemia develops. When SMBG does not meet the established goals, ADA, AACE, and the American College of Obstetricians and Gynecologists (ACOG) concur that insulin is the first-line therapy, either via multiple daily injections or continuous subcutaneous insulin infusion.5,6,12
Regular and neutral protamine hagedorn (NPH) insulin, both of which are classified as pregnancy category B, have been the mainstay therapy. Recently, rapid-acting insulin aspart has been approved for use in pregnancy, and lispro is considered a treatment option for patients with GDM by AACE and the 2008 Expert Review of Obstetrics and Gynecology on the Management of GDM.6,29 Likewise, 70/30 aspart mix and 75/25 lispro mix are now pregnancy category B. For basal insulin, detemir appears to be safe during pregnancy in early studies, while glargine, though used, has no conclusive reports on safety30,31; both remain pregnancy category C.
Recent studies have indicated that pregravid BMI should also be considered by clinicians who treat GDM. Results showed overweight women with GDM that is well controlled on diet alone had a 50% greater risk for delivering a macrosomic infant than did normal-weight patients with GDM, and this risk increased twofold for obese women. Of note, overweight and obese women with GDM that was well controlled on insulin had no increased risk for fetal macrosomia, compared to the reference group.32
The oral medications metformin (pregnancy category B) and glyburide, the most widely studied sulfonylurea (pregnancy category C), have been shown to be effective alternatives to insulin without adverse effects in some women.6 However, the studies have been limited.
Long-term effects on children exposed to these medications in utero are yet to be determined, and randomized, prospective studies using oral diabetic medications in pregnancy, with long-term follow-up of mothers and offspring, are needed. (Consequently, in our practice, we only use insulin therapy for GDM.)
Q: What is the follow-up for women diagnosed with GDM?
Due to the prevalence of T2DM in women with GDM, ADA, AACE, and ACOG concur with the recommendations from the Fifth International Workshop-Conference on Gestational Diabetes that a 75-g, 2-h OGTT be administered six to 12 weeks after delivery in women with GDM who do not have diabetes immediately postpartum.5,6,12,23 Thereafter, ADA recommends resuming routine screening every three years, with more frequent testing depending on initial results and risk status.5 Education regarding diet, weight loss, and exercise should be presented as lifestyle changes and adopted by the entire family, since the infant is also at risk for obesity and the metabolic syndrome.
REFERENCES
1. Metzger BE, Coustan DR. Summary and recommendations of the Fourth International Workshop Conference on Gestational Diabetes Mellitus. Diabetes Care. 1998;21(suppl 2):B161–B167.
2. CDC. National diabetes fact sheet, 2011. www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Accessed May 24, 2011.
3. Metzger BE, Lowe LP, Dyer AR, et al; HAPO Study Cooperative Research Group. Hyperglycemia and adverse pregnancy outcomes. N Engl J Med. 2008;358(19):1991-2002.
4. Metzger BE, Gabbe SG, Persson B, et al; International Association of Diabetes and Pregnancy Study Groups Consensus Panel. International Association of Diabetes and Pregnancy Study Groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care. 2010;33(3):676-682.
5. American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care. 2011;34(suppl 1):S11-S61.
6. Handelsman Y, Mechanick JI, Blonde L, et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for developing a diabetes mellitus comprehensive care plan. Endocr Pract. 2011;17(suppl 2):1-53.
7. Barbour LA, McCurdy CE, Hernandez TL, et al. Cellular mechanisms for insulin resistance in normal pregnancy and gestational diabetes. Diabetes Care. 2007;30(suppl 2):S112-S119.
8. Reece EA, Homko CJ. Why do diabetic women deliver malformed infants? Clin Obstet Gynecol. 2000;43(1):32-45.
9. Cundy T, Gamble G, Townend K, et al. Perinatal mortality in type 2 diabetes mellitus. Diabet Med. 2000;17(1):33-39.
10. Barbour LA. New concepts in insulin resistance of pregnancy and gestational diabetes: long-term implications for mother and offspring. J Obstet Gynaecol. 2003;23(5):545-549.
11. Weiss PA, Scholz HS, Haas J, Tamussino KF. Effect of fetal hyperinsulinism on oral glucose tolerance test results in patients with gestational diabetes mellitus. Am J Obstet Gynecol. 2001;184(3):470-475.
12. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin: clinical management guidelines for obstetrician-gynecologists. Obstet Gynecol. 2001;98(3):525-538.
13. Hillier TA, Pedula KL, Schmidt MM, et al. Childhood obesity and metabolic imprinting: the ongoing effects of maternal hyperglycemia. Diabetes Care. 2007;30(9):2287-2292.
14. Gillman MW, Rifas-Shiman S, Berkey CS, et al. Maternal gestational diabetes, birth weight, and adolescent obesity. Pediatrics. 2003;111(3):e221-e226.
15. Silverman BL, Rizzo TA, Cho NH, Metzger BE. Long-term effects of the intrauterine environment. Diabetes Care. 1998;21(suppl 2):B142- B149.
16. Pettitt DJ, Nelson RG, Saad MF, et al. Diabetes and obesity in the offspring of Pima Indian women with diabetes during pregnancy. Diabetes Care. 1993;16(1):310-314.
17. Catalano PM, Hauguel-de Mouzon S. Is it time to revisit the Pedersen hypothesis in the face of the obesity epidemic? Am J Obstet Gynecol. 2011 Feb 2; [Epub ahead of print].
18. Catalano PM. Obesity, insulin resistance, and pregnancy outcome. Reproduction. 2010;140(3):365-371.
19. Catalano PM, Presley L, Minium J, Hauguel-de Mouzon S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care. 2009;32(6):1076-1080.
20. Catalano PM. Obesity and pregnancy—the propagation of a viscous cycle? J Clin Endocrinol Metab. 2003;88(8):3505-3506.
21. Kim C, Newton KM, Knopp RH. Gestational diabetes and the incidence of type 2 diabetes: a systematic review. Diabetes Care. 2002;25(10):1862-1868.
22. Lawrence JM, Contreras R, Chen W, Sacks DA. Trends in the prevalence of preexisting diabetes and gestational diabetes mellitus among a racially/ethnically diverse population of pregnant women, 1999-2005. Diabetes Care. 2008;31(5):899-904.
23. Metzger BE, Buchanan TA, Coustan DR, et al. Summary and recommendations of the Fifth International Workshop-Conference on Gestational Diabetes Mellitus. Diabetes Care. 2007;30(suppl 2):S251-S260.
24. Kitzmiller JL, Block JM, Brown FM, et al. Managing preexisting diabetes for pregnancy: summary of evidence and consensus recommendations for care. Diabetes Care. 2008;31(5):1060-1079.
25. Landon MB, Spongy CY, Thorn E, et al; Eunice Kennedy Shriver National Institute of Child Health and Human Development Maternal-Fetal Medicine Units Network. A multicenter, randomized trial of treatment for mild gestational diabetes. N Engl J Med. 2009;361(14):1339-1348.
26. Crowther CA, Hiller JE, Moss JR, et al; Australian Carbohydrate Intolerance Study in Pregnant Women (ACHOIS) Trial Group. Effect of treatment of gestational diabetes mellitus on pregnancy outcomes. N Engl J Med. 2005;352(24):2477-2486.
27. Knopp RH, Magee MS, Raisys V, et al. Hypocaloric diets and ketogenesis in the management of obese gestational diabetic women. J Am Coll Nutr. 1991;10(6):649-667.
28. Rizzo T, Metzger BE, Burns WJ, Burns K. Correlations between antepartum maternal metabolism and child intelligence. N Engl J Med. 1991; 325(13):911-916.
29. Menato G, Bo S, Signorile A, et al. Current management of gestational diabetes mellitus. Expert Rev of Obstet Gynecol. 2008;3(1):73-91.
30. Sciacca L, Marotta V, Insalaco F, et al. Use of insulin detemir during pregnancy. Nutr Metab Cardiovasc Dis. 2010;20(4):e15-e16.
31. Lapolla A, Di Cianni G, Bruttomesso D, et al. Use of insulin detemir in pregnancy: a report on 10 type 1 diabetic women. Diabet Med. 2009; 26(11):1181-1182.
32. Langer O, Yogev Y, Xenakis EM, Brustman L. Overweight and obese gestational diabetes: impact on pregnancy outcome. Am J Obstet Gynecol. 2005;192(6):1768-1776.
Q: Why has there been a change in the standards of care for gestational diabetes mellitus?
With the obesity epidemic in this country, a greater number of women in their childbearing years are at risk for this continuum of metabolic syndrome, gestational diabetes mellitus (GDM), and type 2 diabetes mellitus (T2DM). GDM has long been defined as glucose intolerance with onset or first recognition during pregnancy.1 Compounding this definition are concerns that undiagnosed type 1 diabetes mellitus (T1DM) or T2DM may have been present at conception or that glucose intolerance may continue after pregnancy, developing into T2DM postpartum or prior to subsequent pregnancies.
In April 2011, the CDC reported that GDM occurs in 2% to 10% of pregnancies and is more likely in women who have a family history of diabetes, are obese, or are African-American, Hispanic/Latino American, or American Indian.2
Furthermore, in 2008, the Hyperglycemia and Adverse Pregnancy Outcomes (HAPO) study demonstrated a continuous association of adverse pregnancy outcomes (maternal, fetal, and neonatal) with increasing maternal glucose levels, even at glycemic levels previously considered normal.3 In response, the International Association of Diabetes and Pregnancy Study Groups (IADPSG) developed revised recommendations for diagnosing GDM; these were published in 2010.4 Professional organizations such as the American Diabetes Association (ADA) and the American Association of Clinical Endocrinologists (AACE) are now incorporating these revisions into their guidelines.5,6
Q: What causes GDM?
After 20 weeks’ gestation, there is an approximately 50% increase in insulin resistance in all women, most likely resulting from elevated levels of reproductive/placental hormones and possibly cytokines. As levels rise for the duration of the pregnancy, insulin resistance continually increases.
Women with normal pancreatic function are able to secrete the additional insulin required (as much as 200% to 250% by late pregnancy) to maintain euglycemia. Women predisposed to insulin resistance prior to pregnancy have already begun taxing their pancreatic beta-cells for increased production. As insulin resistance increases during the second and third trimesters, they are less likely to meet the additional demands for insulin, with impaired insulin secretion resulting in a greater risk for hyperglycemia.7
Q: What are the risks of maternal hyperglycemia for the child?
Hyperglycemia during the first trimester, either due to uncontrolled T1DM or undiagnosed T2DM, may cause birth defects, including neural tube and cardiac malformations; it can also contribute to early fetal loss.8,9 Maternal hyperglycemia during the second and third trimesters, as seen with the onset of GDM, is also associated with an increased risk for other complications, and HAPO reported that these complications increased with rising glucose levels previously considered normal.4
According to Pedersen’s hypothesis, maternal hyperglycemia transfers across the placenta, inducing fetal hyperglycemia, compensatory fetal hyperinsulinemia, and consequently increased adipose deposition of nutrients, resulting in macrosomia—the most common complication of GDM.10 Fetal hyperinsulinemia can siphon glucose from the mother, known as steal phenomenon, creating the appearance of maternal euglycemia.11 For this reason, ultrasound surveillance is used to monitor for accelerated fetal growth.
An estimated fetal weight of more than 4,500 g carries a high risk for shoulder dystocia with vaginal delivery, so elective cesarean section is usually recommended.12 Common complications in the infant include neonatal hypoglycemia secondary to fetal hyperinsulinemia, polycythemia, hyperbilirubinemia, and an increased need for neonatal intensive care.4 Furthermore, uncontrolled maternal GDM results in offspring at risk for childhood obesity and impaired glucose tolerance, giving rise to the theory of fetal imprinting and perpetuating a vicious cycle.13-16
In light of worldwide weight trends and with obesity considered the number one risk factor for insulin resistance, which is characterized by elevated triglycerides and free fatty acids, Catalano is revisiting Pedersen’s hypothesis, adding that the availability of maternal lipids for fetal lipogenesis is a possible contributing factor to fetal fat accumulation.17 This could explain why pregnancies complicated by obesity carry many of the same risks, or compound the maternal-fetal risks, that are associated with hyperglycemia in pregnancy.18-20
Q: What are the risks of GDM for the mother?
The immediate risks are an increased incidence of cesarean section, preeclampsia, and preterm delivery.3 Since GDM falls within the continuum for developing T2DM, pregnancy serves as a “stress test” in women who are genetically predisposed. Women who develop GDM have about a 50% risk for developing T2DM within five to 10 years.21
Q: How is GDM diagnosed?
As a result of the obesity and diabetes epidemics in this country, increasing numbers of women have undiagnosed T2DM with pregnancy.22 For this reason, the new guidelines recommend using standard diabetes screening for women with risk factors for T2DM at the first prenatal visit. Women who test positive are given a diagnosis of diabetes and should begin diabetes self-management education and therapeutic intervention, when indicated, without delay. AACE recommends that A1C be used as a screening test, due to potential interfering factors not related to glycemia, and that the diagnosis of diabetes be based on plasma glucose readings.6
Using HAPO data, the IADPSG proposed a new set of GDM diagnostic criteria in 2010, which has been adopted by ADA and AACE in 2011.3-6 Following the new standards, all women not previously diagnosed with diabetes should be screened at 24 to 28 weeks’ gestation using a 75-g, 2-h oral glucose tolerance test (OGTT) after an 8-h overnight fast. The diagnosis of GDM is made if there is one abnormal plasma glucose value: fasting glucose ≥ 92 mg/dL, 1-h glucose ≥ 180 mg/dL, or 2-h glucose ≥ 153 mg/dL.
Q: What are the goals for self-monitoring of blood glucose (SMBG) in pregnancy?
HAPO revealed that fasting and postprandial hyperglycemia all predicted risk for macrosomia, fetal hyperinsulinemia, and cesarean delivery.3 ADA and AACE concur with the recommendations from the Fifth International Workshop-Conference on Gestational Diabetes that SMBG goals for women with GDM are as follows: preprandial ≤ 95 mg/dL and either 1-h postprandial ≤ 140 mg/dL or 2-h postprandial ≤ 120 mg/dL.5,6,23
SMBG is recommended at least three times daily for pregnant women taking insulin, but ADA also recommends that monitoring be dictated by the needs and goals of the patient.5 For women with T1DM and T2DM who become pregnant, ADA and AACE concur with a recent consensus statement recommending avoidance of excessive hypoglycemia while aiming for glycemic goals of: preprandial, bedtime, and overnight glucose, 60 to 99 mg/dL, peak postprandial glucose, 100 to 129 mg/dL, and A1C 5,6,24
Q: What is the best therapy for women with GDM?
Although more women will be diagnosed with GDM using the 2011 guidelines, therapeutic lifestyle changes and medical nutrition therapy (diet) will be key elements in their management, as demonstrated in the Maternal-Fetal Medicine Units Network and the Australian Carbohydrate Intolerance Study in Pregnant Women trials.25,26 Women should not arbitrarily restrict calories, as ketosis can result from simply an overnight fast, and maternal ketones have been shown to impair the IQ of offspring.27,28
In many cases, diet alone, while preventing ketonuria, may control postprandial plasma glucose, but diet-control is less successful when fasting hyperglycemia develops. When SMBG does not meet the established goals, ADA, AACE, and the American College of Obstetricians and Gynecologists (ACOG) concur that insulin is the first-line therapy, either via multiple daily injections or continuous subcutaneous insulin infusion.5,6,12
Regular and neutral protamine hagedorn (NPH) insulin, both of which are classified as pregnancy category B, have been the mainstay therapy. Recently, rapid-acting insulin aspart has been approved for use in pregnancy, and lispro is considered a treatment option for patients with GDM by AACE and the 2008 Expert Review of Obstetrics and Gynecology on the Management of GDM.6,29 Likewise, 70/30 aspart mix and 75/25 lispro mix are now pregnancy category B. For basal insulin, detemir appears to be safe during pregnancy in early studies, while glargine, though used, has no conclusive reports on safety30,31; both remain pregnancy category C.
Recent studies have indicated that pregravid BMI should also be considered by clinicians who treat GDM. Results showed overweight women with GDM that is well controlled on diet alone had a 50% greater risk for delivering a macrosomic infant than did normal-weight patients with GDM, and this risk increased twofold for obese women. Of note, overweight and obese women with GDM that was well controlled on insulin had no increased risk for fetal macrosomia, compared to the reference group.32
The oral medications metformin (pregnancy category B) and glyburide, the most widely studied sulfonylurea (pregnancy category C), have been shown to be effective alternatives to insulin without adverse effects in some women.6 However, the studies have been limited.
Long-term effects on children exposed to these medications in utero are yet to be determined, and randomized, prospective studies using oral diabetic medications in pregnancy, with long-term follow-up of mothers and offspring, are needed. (Consequently, in our practice, we only use insulin therapy for GDM.)
Q: What is the follow-up for women diagnosed with GDM?
Due to the prevalence of T2DM in women with GDM, ADA, AACE, and ACOG concur with the recommendations from the Fifth International Workshop-Conference on Gestational Diabetes that a 75-g, 2-h OGTT be administered six to 12 weeks after delivery in women with GDM who do not have diabetes immediately postpartum.5,6,12,23 Thereafter, ADA recommends resuming routine screening every three years, with more frequent testing depending on initial results and risk status.5 Education regarding diet, weight loss, and exercise should be presented as lifestyle changes and adopted by the entire family, since the infant is also at risk for obesity and the metabolic syndrome.
REFERENCES
1. Metzger BE, Coustan DR. Summary and recommendations of the Fourth International Workshop Conference on Gestational Diabetes Mellitus. Diabetes Care. 1998;21(suppl 2):B161–B167.
2. CDC. National diabetes fact sheet, 2011. www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf. Accessed May 24, 2011.
3. Metzger BE, Lowe LP, Dyer AR, et al; HAPO Study Cooperative Research Group. Hyperglycemia and adverse pregnancy outcomes. N Engl J Med. 2008;358(19):1991-2002.
4. Metzger BE, Gabbe SG, Persson B, et al; International Association of Diabetes and Pregnancy Study Groups Consensus Panel. International Association of Diabetes and Pregnancy Study Groups recommendations on the diagnosis and classification of hyperglycemia in pregnancy. Diabetes Care. 2010;33(3):676-682.
5. American Diabetes Association. Standards of medical care in diabetes—2011. Diabetes Care. 2011;34(suppl 1):S11-S61.
6. Handelsman Y, Mechanick JI, Blonde L, et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for developing a diabetes mellitus comprehensive care plan. Endocr Pract. 2011;17(suppl 2):1-53.
7. Barbour LA, McCurdy CE, Hernandez TL, et al. Cellular mechanisms for insulin resistance in normal pregnancy and gestational diabetes. Diabetes Care. 2007;30(suppl 2):S112-S119.
8. Reece EA, Homko CJ. Why do diabetic women deliver malformed infants? Clin Obstet Gynecol. 2000;43(1):32-45.
9. Cundy T, Gamble G, Townend K, et al. Perinatal mortality in type 2 diabetes mellitus. Diabet Med. 2000;17(1):33-39.
10. Barbour LA. New concepts in insulin resistance of pregnancy and gestational diabetes: long-term implications for mother and offspring. J Obstet Gynaecol. 2003;23(5):545-549.
11. Weiss PA, Scholz HS, Haas J, Tamussino KF. Effect of fetal hyperinsulinism on oral glucose tolerance test results in patients with gestational diabetes mellitus. Am J Obstet Gynecol. 2001;184(3):470-475.
12. American College of Obstetricians and Gynecologists. ACOG Practice Bulletin: clinical management guidelines for obstetrician-gynecologists. Obstet Gynecol. 2001;98(3):525-538.
13. Hillier TA, Pedula KL, Schmidt MM, et al. Childhood obesity and metabolic imprinting: the ongoing effects of maternal hyperglycemia. Diabetes Care. 2007;30(9):2287-2292.
14. Gillman MW, Rifas-Shiman S, Berkey CS, et al. Maternal gestational diabetes, birth weight, and adolescent obesity. Pediatrics. 2003;111(3):e221-e226.
15. Silverman BL, Rizzo TA, Cho NH, Metzger BE. Long-term effects of the intrauterine environment. Diabetes Care. 1998;21(suppl 2):B142- B149.
16. Pettitt DJ, Nelson RG, Saad MF, et al. Diabetes and obesity in the offspring of Pima Indian women with diabetes during pregnancy. Diabetes Care. 1993;16(1):310-314.
17. Catalano PM, Hauguel-de Mouzon S. Is it time to revisit the Pedersen hypothesis in the face of the obesity epidemic? Am J Obstet Gynecol. 2011 Feb 2; [Epub ahead of print].
18. Catalano PM. Obesity, insulin resistance, and pregnancy outcome. Reproduction. 2010;140(3):365-371.
19. Catalano PM, Presley L, Minium J, Hauguel-de Mouzon S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care. 2009;32(6):1076-1080.
20. Catalano PM. Obesity and pregnancy—the propagation of a viscous cycle? J Clin Endocrinol Metab. 2003;88(8):3505-3506.
21. Kim C, Newton KM, Knopp RH. Gestational diabetes and the incidence of type 2 diabetes: a systematic review. Diabetes Care. 2002;25(10):1862-1868.
22. Lawrence JM, Contreras R, Chen W, Sacks DA. Trends in the prevalence of preexisting diabetes and gestational diabetes mellitus among a racially/ethnically diverse population of pregnant women, 1999-2005. Diabetes Care. 2008;31(5):899-904.
23. Metzger BE, Buchanan TA, Coustan DR, et al. Summary and recommendations of the Fifth International Workshop-Conference on Gestational Diabetes Mellitus. Diabetes Care. 2007;30(suppl 2):S251-S260.
24. Kitzmiller JL, Block JM, Brown FM, et al. Managing preexisting diabetes for pregnancy: summary of evidence and consensus recommendations for care. Diabetes Care. 2008;31(5):1060-1079.
25. Landon MB, Spongy CY, Thorn E, et al; Eunice Kennedy Shriver National Institute of Child Health and Human Development Maternal-Fetal Medicine Units Network. A multicenter, randomized trial of treatment for mild gestational diabetes. N Engl J Med. 2009;361(14):1339-1348.
26. Crowther CA, Hiller JE, Moss JR, et al; Australian Carbohydrate Intolerance Study in Pregnant Women (ACHOIS) Trial Group. Effect of treatment of gestational diabetes mellitus on pregnancy outcomes. N Engl J Med. 2005;352(24):2477-2486.
27. Knopp RH, Magee MS, Raisys V, et al. Hypocaloric diets and ketogenesis in the management of obese gestational diabetic women. J Am Coll Nutr. 1991;10(6):649-667.
28. Rizzo T, Metzger BE, Burns WJ, Burns K. Correlations between antepartum maternal metabolism and child intelligence. N Engl J Med. 1991; 325(13):911-916.
29. Menato G, Bo S, Signorile A, et al. Current management of gestational diabetes mellitus. Expert Rev of Obstet Gynecol. 2008;3(1):73-91.
30. Sciacca L, Marotta V, Insalaco F, et al. Use of insulin detemir during pregnancy. Nutr Metab Cardiovasc Dis. 2010;20(4):e15-e16.
31. Lapolla A, Di Cianni G, Bruttomesso D, et al. Use of insulin detemir in pregnancy: a report on 10 type 1 diabetic women. Diabet Med. 2009; 26(11):1181-1182.
32. Langer O, Yogev Y, Xenakis EM, Brustman L. Overweight and obese gestational diabetes: impact on pregnancy outcome. Am J Obstet Gynecol. 2005;192(6):1768-1776.