User login
Don't forget non-Alzheimer dementias
Dementia is not always due to Alzheimer disease. An accurate diagnosis is important, as the various causative conditions can differ in their course and treatment.
Dementia refers to cognitive impairment severe enough to interfere with the ability to independently perform activities of daily living. It can occur at any age but is most common after age 60. Some studies estimate that 13.9% of people age 71 and older have some form of dementia.1 The prevalence increases with age, ranging from 5% at age 70 to 79 to 37% at age 90 and older.1
Alzheimer disease accounts for about 60% to 80% of cases,2 or an estimated 4.7 million people age 65 and older in the United States, a number anticipated to climb to 13.8 million by 2050.3
Other types of dementia are less often considered and are challenging to recognize, although many have distinct characteristics. This article summarizes the features and management of the more common non-Alzheimer dementias:
- Vascular dementia
- Dementia with Lewy bodies
- Progressive supranuclear palsy
- Corticobasal degeneration
- Multiple system atrophy
- Parkinson disease dementia
- Frontotemporal dementia
- Primary progressive aphasia
- Normal-pressure hydrocephalus
- Rapidly progressive dementia (ie, Creutzfeld-Jakob disease, autoimmune disease).
VASCULAR DEMENTIA
After Alzheimer disease, vascular dementia is the most common dementia, accounting for about 20% to 30% of cases. Clinical criteria have not been widely accepted, although several have been published, including those in the Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) and the National Institute of Neurological and Communicative Diseases and Stroke-Association Internationale pour la Recherche et l’Enseignement en Neurosciences.
Risk factors for vascular dementia include cerebrovascular disease (hypertension, diabetes, hyperlipidemia) and coexisting conditions related to atherosclerosis (coronary artery disease, peripheral artery disease).
The Hachinski Ischemic Score is a good bedside tool to help differentiate Alzheimer dementia from vascular dementia.5
Sudden onset and stepwise decline
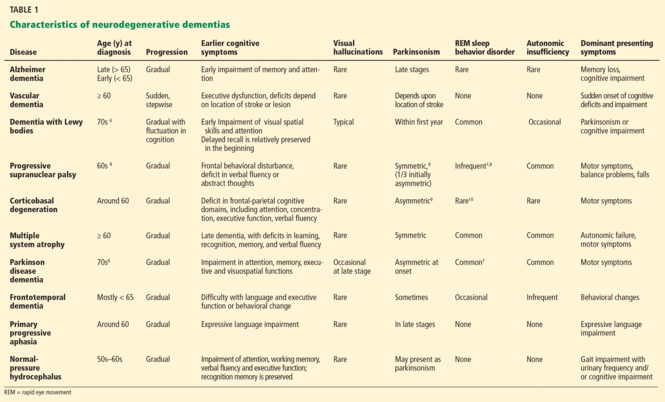
Vascular dementia often presents as a sudden and stepwise progression of cognitive deficits that stabilize and that are caused by vascular insults (Table 1).6–10 Some patients have continuous decline after a vascular event, indicating that Alzheimer dementia may also be present. Dementia is then defined as a mixed type.
Behavioral problems such as physical aggression, hallucinations, paranoia, and mood fluctuations are common.11
Deficits depend on vascular areas affected
Cognitive deficits are heterogeneous and are often related to the location of the vascular insult. Involvement of subcortical areas may result in executive dysfunction, slowed processing speed, and behavioral changes.12
Executive dysfunction may be identified using the Trail Making Test (Part B) or the Executive Interview (EXIT25). Office-based tools such as the Folstein Mini-Mental State Examination, the Montreal Cognitive Assessment, or the St. Louis University Mental Status Examination may also uncover these deficits.
Focal neurologic deficits may be found on clinical examination.
Structural neuroimaging may identify small strokes in areas of the brain affecting cognitive function or occlusion of a larger vessel associated with more profound neurologic deficits. Neuroimaging findings may not correlate with any significant decline noted by the patient, suggesting “silent” strokes.
Treat symptoms and manage risk factors
Although the US Food and Drug Administration (FDA) has not approved any pharmacotherapy for vascular dementia, commonly prescribed cognitive enhancers have demonstrated some benefit.13
Behavioral problems such as aggression can be disturbing to the patient and the caregiver. Nonpharmacologic methods (eg, redirection, rescheduling care activities to avoid conflict, avoiding issues that lead to agitation) should be tried first to address these problems.
Drug therapy may be used off-label for neuropsychiatric symptoms such as hallucinations, delusions, and combativeness, but clinical trials of these agents for this purpose have shown mixed results,14 and their use is often associated with significant risk.15 Antipsychotic drugs are associated with a risk of death and pneumonia when prescribed for dementia. Many also carry a risk of QT prolongation, which is particularly concerning for patients with coronary artery disease or rhythm disturbances.
The key to reducing further decline is to optimize management of vascular risk factors to reduce stroke risk.
DEMENTIA WITH LEWY BODIES
Dementia with Lewy bodies, the next most common neurodegenerative dementia in the elderly, is characterized by progressive loss of cognitive function, prominent visual hallucinations, and parkinsonism (Table 1).6 Disease progression usually occurs over years but can be more rapid than in Alzheimer disease.
Alpha-synucleinopathy results in dysfunction of synaptic vesicles in presynaptic terminals. Lewy bodies may be diffusely spread in cortical and subcortical areas (appearing as spherical masses).
Visual hallucinations are typical

The McKeith criteria16 are the gold standard for diagnosing probable Lewy body dementia, based on clinical and imaging features (Table 2).
Visual hallucinations are usually well formed and detailed. They may initially be pleasant (eg, seeing children and little people) but may evolve to be accompanied by persecutory delusions.
Parkinsonism develops with or after dementia with Lewy bodies
Dementia with Lewy bodies and Parkinson disease dementia share many clinical and pathologic features; Parkinson dementia also is associated with cortical Lewy bodies.
Parkinsonian features include bradykinesia, masked facies, and rigidity. Resting tremor is less common.
The third report of the Dementia With Lewy Bodies Consortium recommends that the condition be diagnosed if dementia occurs before or concurrently with parkinsonism, and dementia with Parkinson disease should be diagnosed if dementia occurs in the context of well-established Parkinson disease.16 The development of dementia within 12 months of extrapyramidal signs suggests dementia with Lewy bodies.
Cognitive deficits fluctuate
Cognitive impairment in Lewy body dementia is characterized by progressive dementia with fluctuations in cognitive performance. Family members or caregivers may report that the patient can carry on a conversation one day and the next day be confused and inattentive. Compared with those with Alzheimer dementia, patients with Lewy body dementia have better delayed recall but more problems with executive functioning (planning) and visuospatial skills (following an unfamiliar route, copying a figure).
Specialized imaging provides clues
Dementia with Lewy bodies is associated with diffuse brain atrophy, with no established characteristic pattern on structural neuroimaging with computed tomography (CT) or magnetic resonance imaging (MRI).17 The contrast agent ioflupane iodine-123 injection (DaTscan) used with single-photon emission CT (SPECT) detects dopamine transporters, which are reduced in parkinsonian syndromes. The scan can also help differentiate between Alzheimer dementia and Lewy body dementia by detecting the loss of functional dopaminergic terminals in the striatum in Lewy body dementia. Alpha-synuclein imaging may become another useful diagnostic tool in the future.
Alzheimer medications may help in dementia with Lewy bodies
Medications with anticholinergic effects and dopamine agonists should be discontinued because of possible effects on cognitive function and parkinsonism. In one clinical trial,18 rivastigmine (Exelon) was found to help cognitive functioning as well as reduce psychotic symptoms in dementia with Lewy bodies, although a recent Cochrane review could not support the evidence for use of all cholinesterase inhibitors in Lewy body dementia.19 In another trial,20 memantine (Namenda) was found to improve global clinical status and behavioral symptoms of Lewy body dementia.
Treating hallucinations of dementia with Lewy bodies
Patients with dementia with Lewy bodies are extremely sensitive to the extrapyramidal side effects of neuroleptic drugs. Some evidence indicates that the atypical antipsychotic drug quetiapine (Seroquel) helps with prominent and disturbing psychotic features and is less likely to worsen parkinsonism than other antipsychotics.21 The best evidence is for clozapine (Clozaril) as a treatment for hallucinations in Parkinson dementia, but the possible side effect of agranulocytosis limits its clinical use. Other atypical antipsychotics such as risperidone (Risperdal) and olanzapine (Zyprexa) are not recommended.22
PROGRESSIVE SUPRANUCLEAR PALSY
Progressive supranuclear palsy is a sporadic atypical parkinsonian disorder with onset between age 50 and 70. Familial cases are infrequent.
Progressive supranuclear palsy presents as early postural instability, vertical supranuclear gaze palsy, and axial muscle rigidity in the first few years. Disease progression is gradual: one study of 50 patients found that the median time from onset to the first key motor impairment (unintelligible speech, no independent walking, inability to stand unassisted, wheelchair-bound, or recommendation for feeding tube placement) was 4 years.23
Histologically, progressive supranuclear palsy is characterized by accumulation of tau protein aggregates in the basal ganglia, brainstem, and cerebral cortex. The degenerative process involves dopaminergic, cholinergic, and gamma-aminobutyric acid (GABA)-ergic neurons.24
Gait and balance problems predominate early in progressive supranuclear palsy
The most commonly used diagnostic criteria are from the National Institute of Neurological Disorders and Stroke. The diagnosis of probable progressive supranuclear palsy requires vertical gaze palsy and falls or the tendency to fall within the first year of disease onset and exclusion of other causes.
The earliest symptom is usually gait and balance impairment.25 Falls (usually backward) and postural instability occur during the first year in 58% of patients.26 Instead of turning en bloc as in Parkinson disease, patients with progressive supranuclear palsy tend to pivot quickly. Patients may also have a coarse groaning voice and moaning. Insomnia has been reported, but rapid-eye-movement sleep behavior disorders are infrequent (unlike in Parkinson disease, multiple system atrophy, and Lewy body dementia).27
Apathy and extreme mood swings
Cognitive impairment is seen in 50% of patients in the early stage of progressive supranuclear palsy. It mostly involves the frontal lobe, including frontal behavioral disturbances (eg, apathy in 91% of patients26 or pseudobulbar affect and extreme emotional lability) and deficits in abstract thoughts or verbal fluency (to test this, patients are asked to say as many words as possible from a category in a given time). Ideomotor apraxia (inability to correctly imitate hand gestures and voluntarily pantomime tool use, such as pretending to brush hair) is rare, despite corticobasal degeneration.28
Vertical gaze palsy
The hallmark of progressive supranuclear palsy is vertical gaze palsy. Initially, this involves slowing of vertical saccades, followed by diminished vertical gaze and more characteristic downward gaze palsy. These findings may develop over 3 to 4 years. Vertical gaze palsy leads to spilling food and tripping while walking.
The gaze abnormality combined with rare blinking and facial dystonia form the classic facial expression of astonishment called “leonine facies.” The face is stiff and deeply furrowed, with a look of surprise.
Axial (especially neck) rigidity is more prominent than limb rigidity. Retrocollis (the head is drawn back) occurs in less than 25% of patients. Parkinsonian features such as bradykinesia affect nearly half of patients by the time of diagnosis.
Instead of the classic symptoms of progressive supranuclear palsy, about one-third of patients present with progressive supranuclear palsy-parkinsonism, which involves asymmetric parkinsonism that initially responds to levodopa.29
MRI shows ‘hummingbird sign’
Brain MRI shows atrophy of the brainstem, particularly the midbrain. Thinning of the superior part of the midbrain and dilation of the third ventricle (“hummingbird sign” on sagittal sections or “morning glory flower” on axial sections) support a diagnosis of progressive supranuclear palsy and differentiate it from Parkinson disease and other atypical parkinsonian disorders.30,31
Levodopa ineffective for supranuclear palsy
There is no treatment to slow progressive supranuclear palsy. Even in high doses, levodopa rarely alleviates parkinsonian features in a clinically meaningful way.26 Successful experimental biologic therapies have been studied in animal models.32 Davunetide is thought to help with neuronal integrity and cell survival through the stabilization of microtubules in preclinical studies, but it has not been used in clinical practice.33
CORTICOBASAL DEGENERATION
Corticobasal degeneration is a progressive, asymmetric movement disorder often manifesting initially with cognitive or behavioral impairment. It is associated with abnormality of the cytoskeleton protein tau. Onset is usually after age 60.
Asymmetric movement disorder with cognitive dysfunction
This diagnosis is clinical. Diagnostic criteria proposed in 2003 include the following core features34:
- Insidious onset and progressive course
- No identifiable cause
- Cortical dysfunction with at least one of the following: apraxia, alien limb phenomenon (one limb moves involuntarily with complex movements, eg, grabbing the other hand), cortical sensory loss, visual hemineglect, nonfluent aphasia
- Extrapyramidal dysfunction: focal rigidity unresponsive to levodopa, asymmetric dystonia.
An international consortium has developed more specific clinical research criteria for probable and possible corticobasal degeneration.35 In a series of 147 patients, the following clinical features were found: parkinsonism (100%), higher cortical dysfunction (93%), dyspraxia (82%), gait disorder (80%), unilateral limb dystonia (71%), tremor (55%), and dementia (25%).36
Behavioral problems commonly include depression; apathy, irritability, and agitation are also reported.37
Cognitive testing may reveal deficits in frontal-parietal cognitive domains including attention and concentration, executive function, verbal fluency, and visuospatial skills.38 Learning disabilities may be improved with verbal cueing (in contrast to Alzheimer disease). Patients may also have impaired graphesthesia (the ability to recognize writing on the skin only by the sensation of touch).39,40
Motor examination may reveal marked asymmetry. Hand, limb, speech, and gait apraxias are common. Gait is typically slow, with short steps and shuffling, and a wide-based or freezing gait. Arm swing may be absent on one side.
Asymmetric cortical atrophy
Early on, MRI may be normal. As the disease progresses, asymmetric cortical atrophy may be seen, especially in the posterior frontal and parietal lobes.
Levodopa ineffective in corticobasal degeneration
Corticobasal degeneration responds poorly to levodopa. Botulinum toxin has been used to help with dystonia and limb pain.
MULTIPLE SYSTEM ATROPHY
Multiple system atrophy is another atypical parkinsonian disorder, most often diagnosed in men over age 60. It is characterized by sporadic parkinsonism, cerebellar signs (involving balance and coordination), pyramidal tract dysfunction, and autonomic insufficiency in varying combinations. Two major subtypes are recognized, depending on whether the predominating presenting features are cerebellar signs or parkinsonism. In contrast to dementia with Lewy bodies, psychiatric symptoms are not a major feature, except possibly depression.41
Diagnosis requires a sporadic progressive disorder that has features of autonomic failure and poor response of parkinsonism or cerebellar ataxia to levodopa.42
Multiple system atrophy is usually not associated with dementia in the early stages, but patients develop deficits in learning, recognition, memory, and verbal fluency as the disease progresses.43 Rapid-eye-movement sleep behavior disorder has been reported in more than half of patients.44
A neurologic examination provides clues
Parkinsonian features are usually symmetric, in contrast to idiopathic Parkinson disease. These signs may include akinesia with rigidity, postural instability, hypokinetic speech, and tremor.
Cerebellar signs include nystagmus and dysarthria (speech disturbance), and gait and limb ataxia.
Pyramidal features include extensor plantar responses and hyperreflexia.
Autonomic dysfunction includes orthostatic hypotension, bladder and rectal atony, loss of sweating, urinary or fecal incontinence, and erectile dysfunction.
Electromyography may demonstrate decreased anal sphincter tone.
MRI shows atrophy of putamen and pons
Brain MRI may show atrophy of the putamen (hypointensity of the putamen with a hyperintense rim). Pons atrophy may also be present, revealing a “hot cross bun” sign in axial images. These combined findings have specificity above 90% but limited sensitivity. These signs are useful to distinguish multiple system atrophy from Parkinson dementia, but their absence does not exclude the diagnosis of multiple system atrophy.45,46
Multiple system atrophy typically responds poorly to levodopa
Levodopa may improve movement and rigidity, but many respond poorly to treatment or lose response after a few years. Fludrocortisone (Florinef) or vasoconstrictors such as midodrine (Orvaten, Proamatine) may help with orthostatic hypotension.47,48
PARKINSON DISEASE DEMENTIA
Dementia eventually develops in most patients with Parkinson disease. Older age and the akinetic rigid form of the disease are associated with higher risk. Diagnosis of idiopathic Parkinson disease before the development of dementia is essential for the diagnosis.
The Movement Disorder Society Task Force has developed new diagnostic criteria.49 Deficits must be present in at least two of the four core cognitive domains (attention, memory, executive, and visuospatial functions) and must be severe enough to affect daily functioning.
Behavioral symptoms such as affective changes, hallucinations, and apathy are common.
MRI shows characteristic brain atrophy in Parkinson disease dementia
MRI shows reduced gray matter volume in the frontal lobe in patients with Parkinson disease without dementia compared with controls. In Parkinson disease dementia, reduced volume extends to temporal, occipital, and subcortical areas. No significant volumetric differences have been observed in Parkinson dementia compared with dementia with Lewy bodies.50 A greater decrease of glucose metabolism has been found in the inferior parietal and occipital lobes in Parkinson disease dementia than in Parkinson disease without dementia.51
Rivastigmine effective for dementia
A Cochrane review supports the use of acetylcholinesterase inhibitors in patients with Parkinson disease dementia, with a positive impact on global assessment, cognitive function, behavioral disturbance, and activities of daily living rating scales.19 At this time, rivastigmine is the only FDA-approved cholinesterase inhibitor for treating Parkinson disease dementia. In clinical trials, memantine did not improve global clinical status or behavioral symptoms of dementia of Parkinson disease.51
FRONTOTEMPORAL DEMENTIA
Frontotemporal dementia frequently starts before age 65 and accounts for 20% to 50% of dementias in this age group.52 Recognition of the condition in older patients is also growing.53 Frontotemporal dementia encompasses a spectrum of dementias, including behavioral variant frontotemporal dementia, semantic dementia, and progressive nonfluent aphasia.54
Gradual onset of uncharacteristic behaviors
Accepted diagnostic criteria include core features of gradual onset, early decline in social and interpersonal conduct, early impairment of self-regulation, emotional blunting, and loss of insight. Many patients are diagnosed with psychiatric conditions. Changes reported by family and caregivers typically deviate substantially from the person’s usual behavior, such as impulsive and inappropriate behaviors or complete withdrawal and apathy.
Language sometimes affected in frontotemporal dementia
Language impairment may be present in some variants. Behavioral and language changes often accompany other forms of dementia (Alzheimer disease, vascular dementia, primary progressive aphasia), making diagnosis more challenging. Office-based testing often does not reveal any deficits, although the Frontal Behavioral Inventory may help.55 A referral to a clinical neuropsychologist may help identify and quantify cognitive impairments.
MRI shows frontotemporal lobes affected
Structural neuroimaging may not reveal abnormalities initially, but with progression, atrophy may be seen in the frontal and temporal lobes. Functional neuroimaging (positron emission tomography, brain SPECT, functional MRI) show hypometabolism in the same areas.
Treat symptoms
There are no specific FDA-approved therapies for frontotemporal dementia. Acetylcholinesterase inhibitors can help progressive nonfluent aphasia in some cases. Selective serotonin reuptake inhibitors may alleviate depressive symptoms, and low doses of atypical antipsychotic medications may help with impulsivity, disinhibition, and aggressive or disruptive behaviors.56
PRIMARY PROGRESSIVE APHASIA
Language impairment predominates
Primary progressive aphasia is a rare form of dementia in which symptoms typically develop around age 60. Pathology is varied. In a study of 60 patients with initial clinical symptoms of primary progressive aphasia, postmortem histology of brain tissue revealed various findings, including those consistent with Alzheimer pathology and motor neuron diseasetype inclusions.57
Patients typically present with expressive language problems as the primary deficit for the first 2 years of the disease, with preservation in other cognitive areas such as memory, visuospatial skills, and executive function.58 Office-based testing may overstate the severity of the dementia, given the dependence of performance on intact language.
It is important to distinguish primary progressive aphasia from other dementias that also affect language. In the frontal variant of frontotemporal dementia, the primary language problem is anomia (inability to name objects) or diminished speech output, which may be accompanied by behavioral problems. Semantic dementia affects word recognition as well as comprehension. In Alzheimer disease, language may be affected along with memory and other areas of cognitive function.
Imaging shows focal degeneration in the left hemisphere
Structural neuroimaging does not initially reveal any deficits, but later it may reveal atrophy in the frontal, perisylvian complex, and temporal areas of the left hemisphere, reflecting the focal nature of the degeneration.59 Functional neuroimaging (positron emission tomography, SPECT) may reveal hypometabolism or diminished blood flow in these areas prior to changes in structural neuroimaging.60
Other communication methods may help
There are no FDA-approved therapies for primary progressive aphasia. Off-label use of some agents (eg, selective serotonin reuptake inhibitors and small doses of antipsychotic medications) has been found useful in small trials.56 Patients may benefit from learning other forms of communication, such as using sign language, laminated cards with printed words or pictures, or artificial voice synthesizers, to express their needs.
NORMAL-PRESSURE HYDROCEPHALUS
Classic triad: Gait, cognition, incontinence
With the onset of symptoms in the sixth or seventh decade, normal-pressure hydrocephalus affects less than 1% of people age 65 and older. It represents up to 5% of dementias, although estimates are influenced by the varied criteria for diagnosis.61 It is characterized by the classic triad of gait impairment, cognitive impairment, and urinary frequency or incontinence.62
Symptoms progress over a period of years, with gait impairment often predominating. As this triad is common in the geriatric population, identifying other explanations is important. Gait impairment caused by spinal stenosis, peripheral neuropathy, or parkinsonism should be explored. Cognitive impairment could be due to depression, Alzheimer disease, or other forms of dementia. Urinary symptoms may be related to detrusor instability or an enlarged prostate.
Gait impairment initially manifests as slowing of gait, but progresses to difficulty with gait initiation. Gait tends to be wide-based (stance more than 1 foot wide).
Cognitive impairment is typically subcortical, manifested as slowed processing speed and impaired executive function. Recall and working memory may be impaired.
Enlarged ventricles seen on imaging in normal-pressure hydrocephalus
Structural neuroimaging reveals enlarged ventricles (Evan’s ratio > 0.358). This can be difficult to distinguish from ventriculomegaly due to cerebral atrophy; assessing the callosal angle on MRI may distinguish the two.63,64 Diagnosis of normal-pressure hydrocephalus can be confirmed using a cerebrospinal fluid infusion test to assess resistance of fluid to resorption.65
Treat with cerebrospinal fluid drainage
Specific tests should be performed to determine candidacy for surgery. These include a high-volume lumbar puncture (40 to 50 mL) or a trial of external lumbar drainage (10 mL per hour for 48 to 72 hours).65 Definitive treatment is surgical placement of a shunt to allow cerebrospinal fluid to drain into the atria or peritoneal cavity.
Surgery may improve gait, but cognitive symptoms often remain,66 and clinical decline may occur after the shunt is placed. Once gait dysfunction is resolved, other explanations for cognitive impairment or residual gait impairment should be considered. An underlying reason for progression of normal-pressure hydrocephalus symptoms after surgical intervention should be identified.67
RAPIDLY PROGRESSIVE DEMENTIAS
Rapidly progressive dementias are among the most challenging of dementing illnesses. They are characterized by a subacute course and an accelerated rate of decline, developing in less than 2 years. Evaluation should typically be more comprehensive than for other types of dementia. The main goal is to diagnose potentially treatable conditions, such as Hashimoto encephalopathy or paraneoplastic limbic encephalitis, and to distinguish these conditions from diseases with a very poor prognosis, such as Creutzfeldt-Jakob disease.
Creutzfeldt-Jakob disease
Creutzfeldt-Jakob disease is a fatal prion-related neurodegenerative illness. Sporadic disease is most common, but variant, familial, and iatrogenic types have been reported. The most common initial symptoms in sporadic disease are cognitive (39%), cerebellar (21%), behavioral (20%), constitutional (20%), sensory (11%), motor (9%), and visual (7%).68
Chronic neurodegenerative diseases can be misdiagnosed as Creutzfeldt-Jakob disease because of an atypical time course and multi-system neurologic findings.

The US Centers for Disease Control and Prevention has adopted criteria for diagnosing probable Creutzfeldt-Jakob disease (Table 3). Routine investigations should also not suggest an alternative diagnosis.69
Autoimmune diseases
Autoimmune conditions may present as a rapidly progressive dementia, including Hashimoto encephalopathy and antibody-mediated limbic encephalitis, either associated with cancer (paraneoplastic) or without cancer (nonparaneoplastic).
Paraneoplastic limbic encephalitis is a group of inflammatory conditions involving antibodies produced within the cerebrospinal fluid and serum resulting in neurologic symptoms. These antibodies react against proteins expressed mostly by a tumor somewhere else in the body.70
Hashimoto encephalitis is a subacute to chronic encephalopathy that may present as dementia with abnormally high levels of thyroid antibodies. The symptoms can vary from confusion to psychosis. There are two main presentations: one involves a relapsing-remitting course with stroke-like episodes (27% of patients) and the second consists of insidious onset of seizures (66% of patients).
Diagnosis involves testing for elevated anti-thyroid peroxidase and thyroglobulin antibodies. MRI findings are nonspecific. Hashimoto encephalitis responds to treatment with corticosteroids, plasmapheresis, or immunosuppressive therapy.71
- Plassman BL, Langa KM, Fisher GG, et al. Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology 2007; 29:125–132.
- Alzheimer’s Association. 2013 Alzheimer’s disease facts and figures. http://www.alz.org/downloads/facts_figures_2013.pdf. Accessed February 3, 2014.
- Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 2013; 80:1778–1783.
- Hou CE, Carlin D, Miller BL. Non-Alzheimer’s disease dementias: anatomic, clinical, and molecular correlates. Can J Psychiatry 2004; 49:164–171.
- Hachinski VC, Iliff LD, Zilhka E, et al. Cerebral blood flow in dementia. Arch Neurol 1975; 32:632–637.
- Fereshtehnejad SM, Religa D, Westman E, Aarsland D, Lökk J, Eriksdotter M. Demography, diagnostics, and medication in dementia with Lewy bodies and Parkinson’s disease with dementia: data from the Swedish Dementia Quality Registry (SveDem). Neuropsychiatr Dis Treat 2013; 9:927–935.
- Nomura T, Inoue Y, Takigawa H, Nakashima K. Comparison of REM sleep behaviour disorder variables between patients with progressive supranuclear palsy and those with Parkinson’s disease. Parkinsonism Relat Disord 2012; 18:394–396.
- Davis PH, Golbe LI, Duvoisin RC, Schoenberg BS. Risk factors for progrssive supranuclear palsy. Neurology 1988; 38:1546–1552.
- Tousi B, Schuele SU, Subramanian T. A 46-year-old woman with rigidity and frequent falls. Cleve Clin J Med 2005; 72:57–63.
- Cooper AD, Josephs KA. Photophobia, visual hallucinations, and REM sleep behavior disorder in progressive supranuclear palsy and corticobasal degeneration: a prospective study. Parkinsonism Relat Disord 2009; 15:59–61.
- Lyketsos CG, Lopez O, Jones B, Fitzpatrick AL, Breitner J, DeKosky S. Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: results from the cardiovascular health study0. JAMA 2002; 288:1475–1483.
- Knopman DS. Dementia and cerebrovascular disease. Mayo Clin Proc 2006; 81:223–230.
- Erkinjuntti T, Kurz A, Gauthier S, Bullock R, Lilienfeld S, Damaraju CV. Efficacy of galantamine in probable vascular dementia and Alzheimer’s disease combined with cerebrovascular disease: a randomised trial. Lancet 2002; 359:1283–1290.
- Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 2005; 293:596–608.
- Schneider LS, Dagerman K, Insel PS. Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebo-controlled trials. Am J Geriatr Psychiatry 2006; 14:191–210.
- McKeith IG, Dickson DW, Lowe J, et al; Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005; 65:1863–1872.
- Tartaglia MC, Rosen HJ, Miller BL. Neuroimaging in dementia. Neurotherapeutics 2011; 8:82–92.
- McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet 2000; 356:2031–2036.
- Rolinski M, Fox C, Maidment I, McShane R. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson’s disease dementia and cognitive impairment in Parkinson’s disease. Cochrane Database Syst Rev 2012; 3:CD006504.
- Emre M, Tsolaki M, Bonucelli U, et al; on behalf of the 11018 Study Investigators. Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010; 9:969–977.
- Kurlan R, Cummings J, Raman R, Thal L; Alzheimer’s Disease Cooperative Study Group. Quetiapine for agitation or psychosis in patients with dementia and parkinsonism. Neurology 2007; 68:1356–1363.
- Ballard C, Aarsland D, Francis P, Corbett A. Neuropsychiatric symptoms in patients with dementias associated with cortical Lewy bodies: pathophysiology, clinical features, and pharmacological management. Drugs Aging 2013; 30:603–611.
- Goetz CG, Leurgans S, Lang AE, Litvan I. Progression of gait, speech and swallowing deficits in progressive supranuclear palsy. Neurology 2003; 60:917–922.
- Kasashima S, Oda Y. Cholinergic neuronal loss in the basal forebrain and mesopontine tegmentum of progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol 2003; 105:117–124.
- Fahn S, Jankovic J, Hallett M, editors. Principles and Practice of Movement Disorders. 2nd ed. New York, NY: Elsevier/Saunders; 2011.
- Litvan I, Mangone CA, McKee A, et al. Natural history of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) and clinical predictors of survival: a clinicopathological study. J Neurol Neurosurg Psychiatry 1996; 60:615–620.
- Boeve BF, Silber MH, Parisi JE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology 2003; 61:40–45.
- Pharr V, Uttl B, Stark M, Litvan I, Fantie B, Grafman J. Comparison of apraxia in corticobasal degeneration and progressive supranuclear palsy. Neurology 2001; 56:957–963.
- Williams DR, Lees AJ. What features improve the accuracy of the clinical diagnosis of progressive supranuclear palsy-parkinsonism (PSP-P)? Mov Disord 2010; 25:357–362.
- Wenning GK, Litvan I, Tolosa E. Milestones in atypical and secondary parkinsonisms. Mov Disord 2011; 26:1083–1095.
- Gallucci M, Limbucci N, Catalucci A, Caulo M. Neurodegenerative diseases. Radiol Clin North Am 2008; 46:799–817.
- Stamelou M, de Silva R, Arias-Carrión O, et al. Rational therapeutic approaches to progressive supranuclear palsy. Brain 2010; 133:1578–1590.
- Gold M, Lorenzl S, Stewart AJ, Morimoto BH, Williams DR, Gozes I. Critical appraisal of the role of davunetide in the treatment of progressive supranuclear palsy. Neuropsychiatr Dis Treat 2012; 8:85–93.
- Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 2003; 54(suppl 5):S15–S19.
- Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013; 80:496–503.
- Kompoliti K, Goetz CG, Boeve BF, et al. Clinical presentation and pharmacological therapy in corticobasal degeneration. Arch Neurol 1998; 55:957–961.
- Litvan I, Cummings JL, Mega M. Neuropsychiatric features of corticobasal degeneration. J Neurol Neurosurg Psychiatry 1998; 65:717–721.
- Pillon B, Blin J, Vidailhet M, et al. The neuropsychological pattern of corticobasal degeneration: comparison with progressive supranuclear palsy and Alzheimer’s disease. Neurology 1995; 45:1477–1483.
- Tang-Wai DF, Josephs KA, Boeve BF, Petersen RC, Parisi JE, Dickson DW. Coexistent Lewy body disease in a case of “visual variant of Alzheimer’s disease.” J Neurol Neurosurg Psychiatry 2003; 74:389.
- Tang-Wai DF, Josephs KA, Boeve BF, Dickson DW, Parisi JE, Petersen RC. Pathologically confirmed corticobasal degeneration presenting with visuospatial dysfunction. Neurology 2003; 61:1134–1135.
- Goto K, Ueki A, Shimode H, Shinjo H, Miwa C, Morita Y. Depression in multiple system atrophy: a case report. Psychiatry Clin Neurosci 2000; 54:507–511.
- Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008; 71:670–676.
- Berent S, Giordani B, Gilman S, et al. Patterns of neuropsychological performance in multiple system atrophy compared to sporadic and hereditary olivopontocerebellar atrophy. Brain Cogn 2002; 50:194–206.
- Ghorayeb I, Yekhlef F, Chrysostome V, Balestre E, Bioulac B, Tison F. Sleep disorders and their determinants in multiple system atrophy. J Neurol Neurosurg Psychiatry 2002; 72:798–800.
- Schrag A, Kingsley D, Phatouros C, et al. Clinical usefulness of magnetic resonance imaging in multiple system atrophy. J Neurol Neurosurg Psychiatry 1998; 65:65–71.
- Massey LA, Micallef C, Paviour DC, et al. Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Mov Disord 2012; 27:1754–1762.
- Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA 1997; 277:1046–1051.
- Mathias CJ, Kimber JR. Postural hypotension: causes, clinical features, investigation, and management. Annu Rev Med 1999; 50:317–336.
- Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord 2007; 22:1689–1707.
- Burton EJ, McKeith IG, Burn DJ, Williams ED, O’Brien JT. Cerebral atrophy in Parkinson’s disease with and without dementia: a comparison with Alzheimer’s disease, dementia with Lewy bodies and controls. Brain 2004; 127:791–800.
- Piert M, Koeppe RA, Giordani B, Minoshima S, Kuhl DE. Determination of regional rate constants from dynamic FDG-PET studies in Parkinson’s disease. J Nucl Med 1996; 37:1115–1122.
- Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology 2002; 58:1615–1621.
- Baborie A, Griffiths TD, Jaros E, et al. Frontotemporal dementia in elderly individuals. Arch Neurol 2012; 69:1052–1960.
- Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998; 51:1546–1554.
- Kertesz A, Nadkarni N, Davidson W, Thomas AW. The Frontal Behavioral Inventory in the differential diagnosis of frontotemporal dementia. J Int Neuropsychol Soc 2000; 6:460–468.
- Mendez MF, Lauterbach EC, Sampson SM; ANPA Committee on Research. An evidence-based review of the psychopathology of frontotemporal dementia: a report of the ANPA Committee on Research. J Neuropsychiatry Clin Neurosci 2008; 20:130–149.
- Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain 2005; 128:1996–2005.
- Mesulam MM. Primary progressive aphasia—a language-based dementia. N Engl J Med 2003; 349:1535–1542.
- Turner RS, Kenyon LC, Trojanowski JQ, Gonatas N, Grossman M. Clinical, neuroimaging, and pathologic features of progressive nonfluent aphasia. Ann Neurol 1996; 39:166–173.
- Abe K, Ukita H, Yanagihara T. Imaging in primary progressive aphasia. Neuroradiology 1997; 39:556–559.
- Trenkwalder C, Schwarz J, Gebhard J, et al. Starnberg trial on epidemiology of parkinsonism and hypertension in the elderly. Prevalence of Parkinson’s disease and related disorders assessed by a door-to-door survey of inhabitants older than 65 years. Arch Neurol 1995; 52:1017–1022.
- Hakim S, Adams RD. The special clinical problem of symptomatic hydrocephalus with normal cerebrospinal fluid pressure. Observations on cerebrospinal fluid hydrodynamics. J Neurol Sci 1965; 2:307–327.
- Vanneste JA. Diagnosis and management of normal-pressure hydrocephalus. J Neurol 2000; 247:5–14.
- Ishii K, Kanda T, Harada A, et al. Clinical impact of the callosal angle in the diagnosis of idiopathic normal pressure hydrocephalus. Eur Radiol 2008; 18:2678–2683.
- Marmarou A, Bergsneider M, Klinge P, Relkin N, Black PM. The value of supplemental prognostic tests for the preoperative assessment of idiopathic normal-pressure hydrocephalus. Neurosurgery 2005; 57(suppl 3):S17–S28.
- Bergsneider M, Miller C, Vespa PM, Hu X. Surgical management of adult hydrocephalus. Neurosurgery 2008; 62(suppl 2):643–659.
- Malm J, Graff-Radford NR, Ishikawa M, et al. Influence of comorbidities in idiopathic normal pressure hydrocephalus—research and clinical care. A report of the ISHCSF task force on comorbidities in INPH. Fluids Barriers CNS 2013; 10:22.
- Rabinovici GD, Wang PN, Levin J, et al. First symptom in sporadic Creutzfeldt-Jakob disease. Neurology 2006; 66:286–287.
- Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 2009; 132:2659–2668.
- Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol 2008; 7:327–340.
- Chong JY, Rowland LP, Utiger RD. Hashimoto encephalopathy: syndrome or myth? Arch Neurol 2003; 60:164–171.
Dementia is not always due to Alzheimer disease. An accurate diagnosis is important, as the various causative conditions can differ in their course and treatment.
Dementia refers to cognitive impairment severe enough to interfere with the ability to independently perform activities of daily living. It can occur at any age but is most common after age 60. Some studies estimate that 13.9% of people age 71 and older have some form of dementia.1 The prevalence increases with age, ranging from 5% at age 70 to 79 to 37% at age 90 and older.1
Alzheimer disease accounts for about 60% to 80% of cases,2 or an estimated 4.7 million people age 65 and older in the United States, a number anticipated to climb to 13.8 million by 2050.3
Other types of dementia are less often considered and are challenging to recognize, although many have distinct characteristics. This article summarizes the features and management of the more common non-Alzheimer dementias:
- Vascular dementia
- Dementia with Lewy bodies
- Progressive supranuclear palsy
- Corticobasal degeneration
- Multiple system atrophy
- Parkinson disease dementia
- Frontotemporal dementia
- Primary progressive aphasia
- Normal-pressure hydrocephalus
- Rapidly progressive dementia (ie, Creutzfeld-Jakob disease, autoimmune disease).
VASCULAR DEMENTIA
After Alzheimer disease, vascular dementia is the most common dementia, accounting for about 20% to 30% of cases. Clinical criteria have not been widely accepted, although several have been published, including those in the Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) and the National Institute of Neurological and Communicative Diseases and Stroke-Association Internationale pour la Recherche et l’Enseignement en Neurosciences.
Risk factors for vascular dementia include cerebrovascular disease (hypertension, diabetes, hyperlipidemia) and coexisting conditions related to atherosclerosis (coronary artery disease, peripheral artery disease).
The Hachinski Ischemic Score is a good bedside tool to help differentiate Alzheimer dementia from vascular dementia.5
Sudden onset and stepwise decline
Vascular dementia often presents as a sudden and stepwise progression of cognitive deficits that stabilize and that are caused by vascular insults (Table 1).6–10 Some patients have continuous decline after a vascular event, indicating that Alzheimer dementia may also be present. Dementia is then defined as a mixed type.
Behavioral problems such as physical aggression, hallucinations, paranoia, and mood fluctuations are common.11
Deficits depend on vascular areas affected
Cognitive deficits are heterogeneous and are often related to the location of the vascular insult. Involvement of subcortical areas may result in executive dysfunction, slowed processing speed, and behavioral changes.12
Executive dysfunction may be identified using the Trail Making Test (Part B) or the Executive Interview (EXIT25). Office-based tools such as the Folstein Mini-Mental State Examination, the Montreal Cognitive Assessment, or the St. Louis University Mental Status Examination may also uncover these deficits.
Focal neurologic deficits may be found on clinical examination.
Structural neuroimaging may identify small strokes in areas of the brain affecting cognitive function or occlusion of a larger vessel associated with more profound neurologic deficits. Neuroimaging findings may not correlate with any significant decline noted by the patient, suggesting “silent” strokes.
Treat symptoms and manage risk factors
Although the US Food and Drug Administration (FDA) has not approved any pharmacotherapy for vascular dementia, commonly prescribed cognitive enhancers have demonstrated some benefit.13
Behavioral problems such as aggression can be disturbing to the patient and the caregiver. Nonpharmacologic methods (eg, redirection, rescheduling care activities to avoid conflict, avoiding issues that lead to agitation) should be tried first to address these problems.
Drug therapy may be used off-label for neuropsychiatric symptoms such as hallucinations, delusions, and combativeness, but clinical trials of these agents for this purpose have shown mixed results,14 and their use is often associated with significant risk.15 Antipsychotic drugs are associated with a risk of death and pneumonia when prescribed for dementia. Many also carry a risk of QT prolongation, which is particularly concerning for patients with coronary artery disease or rhythm disturbances.
The key to reducing further decline is to optimize management of vascular risk factors to reduce stroke risk.
DEMENTIA WITH LEWY BODIES
Dementia with Lewy bodies, the next most common neurodegenerative dementia in the elderly, is characterized by progressive loss of cognitive function, prominent visual hallucinations, and parkinsonism (Table 1).6 Disease progression usually occurs over years but can be more rapid than in Alzheimer disease.
Alpha-synucleinopathy results in dysfunction of synaptic vesicles in presynaptic terminals. Lewy bodies may be diffusely spread in cortical and subcortical areas (appearing as spherical masses).
Visual hallucinations are typical
The McKeith criteria16 are the gold standard for diagnosing probable Lewy body dementia, based on clinical and imaging features (Table 2).
Visual hallucinations are usually well formed and detailed. They may initially be pleasant (eg, seeing children and little people) but may evolve to be accompanied by persecutory delusions.
Parkinsonism develops with or after dementia with Lewy bodies
Dementia with Lewy bodies and Parkinson disease dementia share many clinical and pathologic features; Parkinson dementia also is associated with cortical Lewy bodies.
Parkinsonian features include bradykinesia, masked facies, and rigidity. Resting tremor is less common.
The third report of the Dementia With Lewy Bodies Consortium recommends that the condition be diagnosed if dementia occurs before or concurrently with parkinsonism, and dementia with Parkinson disease should be diagnosed if dementia occurs in the context of well-established Parkinson disease.16 The development of dementia within 12 months of extrapyramidal signs suggests dementia with Lewy bodies.
Cognitive deficits fluctuate
Cognitive impairment in Lewy body dementia is characterized by progressive dementia with fluctuations in cognitive performance. Family members or caregivers may report that the patient can carry on a conversation one day and the next day be confused and inattentive. Compared with those with Alzheimer dementia, patients with Lewy body dementia have better delayed recall but more problems with executive functioning (planning) and visuospatial skills (following an unfamiliar route, copying a figure).
Specialized imaging provides clues
Dementia with Lewy bodies is associated with diffuse brain atrophy, with no established characteristic pattern on structural neuroimaging with computed tomography (CT) or magnetic resonance imaging (MRI).17 The contrast agent ioflupane iodine-123 injection (DaTscan) used with single-photon emission CT (SPECT) detects dopamine transporters, which are reduced in parkinsonian syndromes. The scan can also help differentiate between Alzheimer dementia and Lewy body dementia by detecting the loss of functional dopaminergic terminals in the striatum in Lewy body dementia. Alpha-synuclein imaging may become another useful diagnostic tool in the future.
Alzheimer medications may help in dementia with Lewy bodies
Medications with anticholinergic effects and dopamine agonists should be discontinued because of possible effects on cognitive function and parkinsonism. In one clinical trial,18 rivastigmine (Exelon) was found to help cognitive functioning as well as reduce psychotic symptoms in dementia with Lewy bodies, although a recent Cochrane review could not support the evidence for use of all cholinesterase inhibitors in Lewy body dementia.19 In another trial,20 memantine (Namenda) was found to improve global clinical status and behavioral symptoms of Lewy body dementia.
Treating hallucinations of dementia with Lewy bodies
Patients with dementia with Lewy bodies are extremely sensitive to the extrapyramidal side effects of neuroleptic drugs. Some evidence indicates that the atypical antipsychotic drug quetiapine (Seroquel) helps with prominent and disturbing psychotic features and is less likely to worsen parkinsonism than other antipsychotics.21 The best evidence is for clozapine (Clozaril) as a treatment for hallucinations in Parkinson dementia, but the possible side effect of agranulocytosis limits its clinical use. Other atypical antipsychotics such as risperidone (Risperdal) and olanzapine (Zyprexa) are not recommended.22
PROGRESSIVE SUPRANUCLEAR PALSY
Progressive supranuclear palsy is a sporadic atypical parkinsonian disorder with onset between age 50 and 70. Familial cases are infrequent.
Progressive supranuclear palsy presents as early postural instability, vertical supranuclear gaze palsy, and axial muscle rigidity in the first few years. Disease progression is gradual: one study of 50 patients found that the median time from onset to the first key motor impairment (unintelligible speech, no independent walking, inability to stand unassisted, wheelchair-bound, or recommendation for feeding tube placement) was 4 years.23
Histologically, progressive supranuclear palsy is characterized by accumulation of tau protein aggregates in the basal ganglia, brainstem, and cerebral cortex. The degenerative process involves dopaminergic, cholinergic, and gamma-aminobutyric acid (GABA)-ergic neurons.24
Gait and balance problems predominate early in progressive supranuclear palsy
The most commonly used diagnostic criteria are from the National Institute of Neurological Disorders and Stroke. The diagnosis of probable progressive supranuclear palsy requires vertical gaze palsy and falls or the tendency to fall within the first year of disease onset and exclusion of other causes.
The earliest symptom is usually gait and balance impairment.25 Falls (usually backward) and postural instability occur during the first year in 58% of patients.26 Instead of turning en bloc as in Parkinson disease, patients with progressive supranuclear palsy tend to pivot quickly. Patients may also have a coarse groaning voice and moaning. Insomnia has been reported, but rapid-eye-movement sleep behavior disorders are infrequent (unlike in Parkinson disease, multiple system atrophy, and Lewy body dementia).27
Apathy and extreme mood swings
Cognitive impairment is seen in 50% of patients in the early stage of progressive supranuclear palsy. It mostly involves the frontal lobe, including frontal behavioral disturbances (eg, apathy in 91% of patients26 or pseudobulbar affect and extreme emotional lability) and deficits in abstract thoughts or verbal fluency (to test this, patients are asked to say as many words as possible from a category in a given time). Ideomotor apraxia (inability to correctly imitate hand gestures and voluntarily pantomime tool use, such as pretending to brush hair) is rare, despite corticobasal degeneration.28
Vertical gaze palsy
The hallmark of progressive supranuclear palsy is vertical gaze palsy. Initially, this involves slowing of vertical saccades, followed by diminished vertical gaze and more characteristic downward gaze palsy. These findings may develop over 3 to 4 years. Vertical gaze palsy leads to spilling food and tripping while walking.
The gaze abnormality combined with rare blinking and facial dystonia form the classic facial expression of astonishment called “leonine facies.” The face is stiff and deeply furrowed, with a look of surprise.
Axial (especially neck) rigidity is more prominent than limb rigidity. Retrocollis (the head is drawn back) occurs in less than 25% of patients. Parkinsonian features such as bradykinesia affect nearly half of patients by the time of diagnosis.
Instead of the classic symptoms of progressive supranuclear palsy, about one-third of patients present with progressive supranuclear palsy-parkinsonism, which involves asymmetric parkinsonism that initially responds to levodopa.29
MRI shows ‘hummingbird sign’
Brain MRI shows atrophy of the brainstem, particularly the midbrain. Thinning of the superior part of the midbrain and dilation of the third ventricle (“hummingbird sign” on sagittal sections or “morning glory flower” on axial sections) support a diagnosis of progressive supranuclear palsy and differentiate it from Parkinson disease and other atypical parkinsonian disorders.30,31
Levodopa ineffective for supranuclear palsy
There is no treatment to slow progressive supranuclear palsy. Even in high doses, levodopa rarely alleviates parkinsonian features in a clinically meaningful way.26 Successful experimental biologic therapies have been studied in animal models.32 Davunetide is thought to help with neuronal integrity and cell survival through the stabilization of microtubules in preclinical studies, but it has not been used in clinical practice.33
CORTICOBASAL DEGENERATION
Corticobasal degeneration is a progressive, asymmetric movement disorder often manifesting initially with cognitive or behavioral impairment. It is associated with abnormality of the cytoskeleton protein tau. Onset is usually after age 60.
Asymmetric movement disorder with cognitive dysfunction
This diagnosis is clinical. Diagnostic criteria proposed in 2003 include the following core features34:
- Insidious onset and progressive course
- No identifiable cause
- Cortical dysfunction with at least one of the following: apraxia, alien limb phenomenon (one limb moves involuntarily with complex movements, eg, grabbing the other hand), cortical sensory loss, visual hemineglect, nonfluent aphasia
- Extrapyramidal dysfunction: focal rigidity unresponsive to levodopa, asymmetric dystonia.
An international consortium has developed more specific clinical research criteria for probable and possible corticobasal degeneration.35 In a series of 147 patients, the following clinical features were found: parkinsonism (100%), higher cortical dysfunction (93%), dyspraxia (82%), gait disorder (80%), unilateral limb dystonia (71%), tremor (55%), and dementia (25%).36
Behavioral problems commonly include depression; apathy, irritability, and agitation are also reported.37
Cognitive testing may reveal deficits in frontal-parietal cognitive domains including attention and concentration, executive function, verbal fluency, and visuospatial skills.38 Learning disabilities may be improved with verbal cueing (in contrast to Alzheimer disease). Patients may also have impaired graphesthesia (the ability to recognize writing on the skin only by the sensation of touch).39,40
Motor examination may reveal marked asymmetry. Hand, limb, speech, and gait apraxias are common. Gait is typically slow, with short steps and shuffling, and a wide-based or freezing gait. Arm swing may be absent on one side.
Asymmetric cortical atrophy
Early on, MRI may be normal. As the disease progresses, asymmetric cortical atrophy may be seen, especially in the posterior frontal and parietal lobes.
Levodopa ineffective in corticobasal degeneration
Corticobasal degeneration responds poorly to levodopa. Botulinum toxin has been used to help with dystonia and limb pain.
MULTIPLE SYSTEM ATROPHY
Multiple system atrophy is another atypical parkinsonian disorder, most often diagnosed in men over age 60. It is characterized by sporadic parkinsonism, cerebellar signs (involving balance and coordination), pyramidal tract dysfunction, and autonomic insufficiency in varying combinations. Two major subtypes are recognized, depending on whether the predominating presenting features are cerebellar signs or parkinsonism. In contrast to dementia with Lewy bodies, psychiatric symptoms are not a major feature, except possibly depression.41
Diagnosis requires a sporadic progressive disorder that has features of autonomic failure and poor response of parkinsonism or cerebellar ataxia to levodopa.42
Multiple system atrophy is usually not associated with dementia in the early stages, but patients develop deficits in learning, recognition, memory, and verbal fluency as the disease progresses.43 Rapid-eye-movement sleep behavior disorder has been reported in more than half of patients.44
A neurologic examination provides clues
Parkinsonian features are usually symmetric, in contrast to idiopathic Parkinson disease. These signs may include akinesia with rigidity, postural instability, hypokinetic speech, and tremor.
Cerebellar signs include nystagmus and dysarthria (speech disturbance), and gait and limb ataxia.
Pyramidal features include extensor plantar responses and hyperreflexia.
Autonomic dysfunction includes orthostatic hypotension, bladder and rectal atony, loss of sweating, urinary or fecal incontinence, and erectile dysfunction.
Electromyography may demonstrate decreased anal sphincter tone.
MRI shows atrophy of putamen and pons
Brain MRI may show atrophy of the putamen (hypointensity of the putamen with a hyperintense rim). Pons atrophy may also be present, revealing a “hot cross bun” sign in axial images. These combined findings have specificity above 90% but limited sensitivity. These signs are useful to distinguish multiple system atrophy from Parkinson dementia, but their absence does not exclude the diagnosis of multiple system atrophy.45,46
Multiple system atrophy typically responds poorly to levodopa
Levodopa may improve movement and rigidity, but many respond poorly to treatment or lose response after a few years. Fludrocortisone (Florinef) or vasoconstrictors such as midodrine (Orvaten, Proamatine) may help with orthostatic hypotension.47,48
PARKINSON DISEASE DEMENTIA
Dementia eventually develops in most patients with Parkinson disease. Older age and the akinetic rigid form of the disease are associated with higher risk. Diagnosis of idiopathic Parkinson disease before the development of dementia is essential for the diagnosis.
The Movement Disorder Society Task Force has developed new diagnostic criteria.49 Deficits must be present in at least two of the four core cognitive domains (attention, memory, executive, and visuospatial functions) and must be severe enough to affect daily functioning.
Behavioral symptoms such as affective changes, hallucinations, and apathy are common.
MRI shows characteristic brain atrophy in Parkinson disease dementia
MRI shows reduced gray matter volume in the frontal lobe in patients with Parkinson disease without dementia compared with controls. In Parkinson disease dementia, reduced volume extends to temporal, occipital, and subcortical areas. No significant volumetric differences have been observed in Parkinson dementia compared with dementia with Lewy bodies.50 A greater decrease of glucose metabolism has been found in the inferior parietal and occipital lobes in Parkinson disease dementia than in Parkinson disease without dementia.51
Rivastigmine effective for dementia
A Cochrane review supports the use of acetylcholinesterase inhibitors in patients with Parkinson disease dementia, with a positive impact on global assessment, cognitive function, behavioral disturbance, and activities of daily living rating scales.19 At this time, rivastigmine is the only FDA-approved cholinesterase inhibitor for treating Parkinson disease dementia. In clinical trials, memantine did not improve global clinical status or behavioral symptoms of dementia of Parkinson disease.51
FRONTOTEMPORAL DEMENTIA
Frontotemporal dementia frequently starts before age 65 and accounts for 20% to 50% of dementias in this age group.52 Recognition of the condition in older patients is also growing.53 Frontotemporal dementia encompasses a spectrum of dementias, including behavioral variant frontotemporal dementia, semantic dementia, and progressive nonfluent aphasia.54
Gradual onset of uncharacteristic behaviors
Accepted diagnostic criteria include core features of gradual onset, early decline in social and interpersonal conduct, early impairment of self-regulation, emotional blunting, and loss of insight. Many patients are diagnosed with psychiatric conditions. Changes reported by family and caregivers typically deviate substantially from the person’s usual behavior, such as impulsive and inappropriate behaviors or complete withdrawal and apathy.
Language sometimes affected in frontotemporal dementia
Language impairment may be present in some variants. Behavioral and language changes often accompany other forms of dementia (Alzheimer disease, vascular dementia, primary progressive aphasia), making diagnosis more challenging. Office-based testing often does not reveal any deficits, although the Frontal Behavioral Inventory may help.55 A referral to a clinical neuropsychologist may help identify and quantify cognitive impairments.
MRI shows frontotemporal lobes affected
Structural neuroimaging may not reveal abnormalities initially, but with progression, atrophy may be seen in the frontal and temporal lobes. Functional neuroimaging (positron emission tomography, brain SPECT, functional MRI) show hypometabolism in the same areas.
Treat symptoms
There are no specific FDA-approved therapies for frontotemporal dementia. Acetylcholinesterase inhibitors can help progressive nonfluent aphasia in some cases. Selective serotonin reuptake inhibitors may alleviate depressive symptoms, and low doses of atypical antipsychotic medications may help with impulsivity, disinhibition, and aggressive or disruptive behaviors.56
PRIMARY PROGRESSIVE APHASIA
Language impairment predominates
Primary progressive aphasia is a rare form of dementia in which symptoms typically develop around age 60. Pathology is varied. In a study of 60 patients with initial clinical symptoms of primary progressive aphasia, postmortem histology of brain tissue revealed various findings, including those consistent with Alzheimer pathology and motor neuron diseasetype inclusions.57
Patients typically present with expressive language problems as the primary deficit for the first 2 years of the disease, with preservation in other cognitive areas such as memory, visuospatial skills, and executive function.58 Office-based testing may overstate the severity of the dementia, given the dependence of performance on intact language.
It is important to distinguish primary progressive aphasia from other dementias that also affect language. In the frontal variant of frontotemporal dementia, the primary language problem is anomia (inability to name objects) or diminished speech output, which may be accompanied by behavioral problems. Semantic dementia affects word recognition as well as comprehension. In Alzheimer disease, language may be affected along with memory and other areas of cognitive function.
Imaging shows focal degeneration in the left hemisphere
Structural neuroimaging does not initially reveal any deficits, but later it may reveal atrophy in the frontal, perisylvian complex, and temporal areas of the left hemisphere, reflecting the focal nature of the degeneration.59 Functional neuroimaging (positron emission tomography, SPECT) may reveal hypometabolism or diminished blood flow in these areas prior to changes in structural neuroimaging.60
Other communication methods may help
There are no FDA-approved therapies for primary progressive aphasia. Off-label use of some agents (eg, selective serotonin reuptake inhibitors and small doses of antipsychotic medications) has been found useful in small trials.56 Patients may benefit from learning other forms of communication, such as using sign language, laminated cards with printed words or pictures, or artificial voice synthesizers, to express their needs.
NORMAL-PRESSURE HYDROCEPHALUS
Classic triad: Gait, cognition, incontinence
With the onset of symptoms in the sixth or seventh decade, normal-pressure hydrocephalus affects less than 1% of people age 65 and older. It represents up to 5% of dementias, although estimates are influenced by the varied criteria for diagnosis.61 It is characterized by the classic triad of gait impairment, cognitive impairment, and urinary frequency or incontinence.62
Symptoms progress over a period of years, with gait impairment often predominating. As this triad is common in the geriatric population, identifying other explanations is important. Gait impairment caused by spinal stenosis, peripheral neuropathy, or parkinsonism should be explored. Cognitive impairment could be due to depression, Alzheimer disease, or other forms of dementia. Urinary symptoms may be related to detrusor instability or an enlarged prostate.
Gait impairment initially manifests as slowing of gait, but progresses to difficulty with gait initiation. Gait tends to be wide-based (stance more than 1 foot wide).
Cognitive impairment is typically subcortical, manifested as slowed processing speed and impaired executive function. Recall and working memory may be impaired.
Enlarged ventricles seen on imaging in normal-pressure hydrocephalus
Structural neuroimaging reveals enlarged ventricles (Evan’s ratio > 0.358). This can be difficult to distinguish from ventriculomegaly due to cerebral atrophy; assessing the callosal angle on MRI may distinguish the two.63,64 Diagnosis of normal-pressure hydrocephalus can be confirmed using a cerebrospinal fluid infusion test to assess resistance of fluid to resorption.65
Treat with cerebrospinal fluid drainage
Specific tests should be performed to determine candidacy for surgery. These include a high-volume lumbar puncture (40 to 50 mL) or a trial of external lumbar drainage (10 mL per hour for 48 to 72 hours).65 Definitive treatment is surgical placement of a shunt to allow cerebrospinal fluid to drain into the atria or peritoneal cavity.
Surgery may improve gait, but cognitive symptoms often remain,66 and clinical decline may occur after the shunt is placed. Once gait dysfunction is resolved, other explanations for cognitive impairment or residual gait impairment should be considered. An underlying reason for progression of normal-pressure hydrocephalus symptoms after surgical intervention should be identified.67
RAPIDLY PROGRESSIVE DEMENTIAS
Rapidly progressive dementias are among the most challenging of dementing illnesses. They are characterized by a subacute course and an accelerated rate of decline, developing in less than 2 years. Evaluation should typically be more comprehensive than for other types of dementia. The main goal is to diagnose potentially treatable conditions, such as Hashimoto encephalopathy or paraneoplastic limbic encephalitis, and to distinguish these conditions from diseases with a very poor prognosis, such as Creutzfeldt-Jakob disease.
Creutzfeldt-Jakob disease
Creutzfeldt-Jakob disease is a fatal prion-related neurodegenerative illness. Sporadic disease is most common, but variant, familial, and iatrogenic types have been reported. The most common initial symptoms in sporadic disease are cognitive (39%), cerebellar (21%), behavioral (20%), constitutional (20%), sensory (11%), motor (9%), and visual (7%).68
Chronic neurodegenerative diseases can be misdiagnosed as Creutzfeldt-Jakob disease because of an atypical time course and multi-system neurologic findings.
The US Centers for Disease Control and Prevention has adopted criteria for diagnosing probable Creutzfeldt-Jakob disease (Table 3). Routine investigations should also not suggest an alternative diagnosis.69
Autoimmune diseases
Autoimmune conditions may present as a rapidly progressive dementia, including Hashimoto encephalopathy and antibody-mediated limbic encephalitis, either associated with cancer (paraneoplastic) or without cancer (nonparaneoplastic).
Paraneoplastic limbic encephalitis is a group of inflammatory conditions involving antibodies produced within the cerebrospinal fluid and serum resulting in neurologic symptoms. These antibodies react against proteins expressed mostly by a tumor somewhere else in the body.70
Hashimoto encephalitis is a subacute to chronic encephalopathy that may present as dementia with abnormally high levels of thyroid antibodies. The symptoms can vary from confusion to psychosis. There are two main presentations: one involves a relapsing-remitting course with stroke-like episodes (27% of patients) and the second consists of insidious onset of seizures (66% of patients).
Diagnosis involves testing for elevated anti-thyroid peroxidase and thyroglobulin antibodies. MRI findings are nonspecific. Hashimoto encephalitis responds to treatment with corticosteroids, plasmapheresis, or immunosuppressive therapy.71
Dementia is not always due to Alzheimer disease. An accurate diagnosis is important, as the various causative conditions can differ in their course and treatment.
Dementia refers to cognitive impairment severe enough to interfere with the ability to independently perform activities of daily living. It can occur at any age but is most common after age 60. Some studies estimate that 13.9% of people age 71 and older have some form of dementia.1 The prevalence increases with age, ranging from 5% at age 70 to 79 to 37% at age 90 and older.1
Alzheimer disease accounts for about 60% to 80% of cases,2 or an estimated 4.7 million people age 65 and older in the United States, a number anticipated to climb to 13.8 million by 2050.3
Other types of dementia are less often considered and are challenging to recognize, although many have distinct characteristics. This article summarizes the features and management of the more common non-Alzheimer dementias:
- Vascular dementia
- Dementia with Lewy bodies
- Progressive supranuclear palsy
- Corticobasal degeneration
- Multiple system atrophy
- Parkinson disease dementia
- Frontotemporal dementia
- Primary progressive aphasia
- Normal-pressure hydrocephalus
- Rapidly progressive dementia (ie, Creutzfeld-Jakob disease, autoimmune disease).
VASCULAR DEMENTIA
After Alzheimer disease, vascular dementia is the most common dementia, accounting for about 20% to 30% of cases. Clinical criteria have not been widely accepted, although several have been published, including those in the Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) and the National Institute of Neurological and Communicative Diseases and Stroke-Association Internationale pour la Recherche et l’Enseignement en Neurosciences.
Risk factors for vascular dementia include cerebrovascular disease (hypertension, diabetes, hyperlipidemia) and coexisting conditions related to atherosclerosis (coronary artery disease, peripheral artery disease).
The Hachinski Ischemic Score is a good bedside tool to help differentiate Alzheimer dementia from vascular dementia.5
Sudden onset and stepwise decline
Vascular dementia often presents as a sudden and stepwise progression of cognitive deficits that stabilize and that are caused by vascular insults (Table 1).6–10 Some patients have continuous decline after a vascular event, indicating that Alzheimer dementia may also be present. Dementia is then defined as a mixed type.
Behavioral problems such as physical aggression, hallucinations, paranoia, and mood fluctuations are common.11
Deficits depend on vascular areas affected
Cognitive deficits are heterogeneous and are often related to the location of the vascular insult. Involvement of subcortical areas may result in executive dysfunction, slowed processing speed, and behavioral changes.12
Executive dysfunction may be identified using the Trail Making Test (Part B) or the Executive Interview (EXIT25). Office-based tools such as the Folstein Mini-Mental State Examination, the Montreal Cognitive Assessment, or the St. Louis University Mental Status Examination may also uncover these deficits.
Focal neurologic deficits may be found on clinical examination.
Structural neuroimaging may identify small strokes in areas of the brain affecting cognitive function or occlusion of a larger vessel associated with more profound neurologic deficits. Neuroimaging findings may not correlate with any significant decline noted by the patient, suggesting “silent” strokes.
Treat symptoms and manage risk factors
Although the US Food and Drug Administration (FDA) has not approved any pharmacotherapy for vascular dementia, commonly prescribed cognitive enhancers have demonstrated some benefit.13
Behavioral problems such as aggression can be disturbing to the patient and the caregiver. Nonpharmacologic methods (eg, redirection, rescheduling care activities to avoid conflict, avoiding issues that lead to agitation) should be tried first to address these problems.
Drug therapy may be used off-label for neuropsychiatric symptoms such as hallucinations, delusions, and combativeness, but clinical trials of these agents for this purpose have shown mixed results,14 and their use is often associated with significant risk.15 Antipsychotic drugs are associated with a risk of death and pneumonia when prescribed for dementia. Many also carry a risk of QT prolongation, which is particularly concerning for patients with coronary artery disease or rhythm disturbances.
The key to reducing further decline is to optimize management of vascular risk factors to reduce stroke risk.
DEMENTIA WITH LEWY BODIES
Dementia with Lewy bodies, the next most common neurodegenerative dementia in the elderly, is characterized by progressive loss of cognitive function, prominent visual hallucinations, and parkinsonism (Table 1).6 Disease progression usually occurs over years but can be more rapid than in Alzheimer disease.
Alpha-synucleinopathy results in dysfunction of synaptic vesicles in presynaptic terminals. Lewy bodies may be diffusely spread in cortical and subcortical areas (appearing as spherical masses).
Visual hallucinations are typical
The McKeith criteria16 are the gold standard for diagnosing probable Lewy body dementia, based on clinical and imaging features (Table 2).
Visual hallucinations are usually well formed and detailed. They may initially be pleasant (eg, seeing children and little people) but may evolve to be accompanied by persecutory delusions.
Parkinsonism develops with or after dementia with Lewy bodies
Dementia with Lewy bodies and Parkinson disease dementia share many clinical and pathologic features; Parkinson dementia also is associated with cortical Lewy bodies.
Parkinsonian features include bradykinesia, masked facies, and rigidity. Resting tremor is less common.
The third report of the Dementia With Lewy Bodies Consortium recommends that the condition be diagnosed if dementia occurs before or concurrently with parkinsonism, and dementia with Parkinson disease should be diagnosed if dementia occurs in the context of well-established Parkinson disease.16 The development of dementia within 12 months of extrapyramidal signs suggests dementia with Lewy bodies.
Cognitive deficits fluctuate
Cognitive impairment in Lewy body dementia is characterized by progressive dementia with fluctuations in cognitive performance. Family members or caregivers may report that the patient can carry on a conversation one day and the next day be confused and inattentive. Compared with those with Alzheimer dementia, patients with Lewy body dementia have better delayed recall but more problems with executive functioning (planning) and visuospatial skills (following an unfamiliar route, copying a figure).
Specialized imaging provides clues
Dementia with Lewy bodies is associated with diffuse brain atrophy, with no established characteristic pattern on structural neuroimaging with computed tomography (CT) or magnetic resonance imaging (MRI).17 The contrast agent ioflupane iodine-123 injection (DaTscan) used with single-photon emission CT (SPECT) detects dopamine transporters, which are reduced in parkinsonian syndromes. The scan can also help differentiate between Alzheimer dementia and Lewy body dementia by detecting the loss of functional dopaminergic terminals in the striatum in Lewy body dementia. Alpha-synuclein imaging may become another useful diagnostic tool in the future.
Alzheimer medications may help in dementia with Lewy bodies
Medications with anticholinergic effects and dopamine agonists should be discontinued because of possible effects on cognitive function and parkinsonism. In one clinical trial,18 rivastigmine (Exelon) was found to help cognitive functioning as well as reduce psychotic symptoms in dementia with Lewy bodies, although a recent Cochrane review could not support the evidence for use of all cholinesterase inhibitors in Lewy body dementia.19 In another trial,20 memantine (Namenda) was found to improve global clinical status and behavioral symptoms of Lewy body dementia.
Treating hallucinations of dementia with Lewy bodies
Patients with dementia with Lewy bodies are extremely sensitive to the extrapyramidal side effects of neuroleptic drugs. Some evidence indicates that the atypical antipsychotic drug quetiapine (Seroquel) helps with prominent and disturbing psychotic features and is less likely to worsen parkinsonism than other antipsychotics.21 The best evidence is for clozapine (Clozaril) as a treatment for hallucinations in Parkinson dementia, but the possible side effect of agranulocytosis limits its clinical use. Other atypical antipsychotics such as risperidone (Risperdal) and olanzapine (Zyprexa) are not recommended.22
PROGRESSIVE SUPRANUCLEAR PALSY
Progressive supranuclear palsy is a sporadic atypical parkinsonian disorder with onset between age 50 and 70. Familial cases are infrequent.
Progressive supranuclear palsy presents as early postural instability, vertical supranuclear gaze palsy, and axial muscle rigidity in the first few years. Disease progression is gradual: one study of 50 patients found that the median time from onset to the first key motor impairment (unintelligible speech, no independent walking, inability to stand unassisted, wheelchair-bound, or recommendation for feeding tube placement) was 4 years.23
Histologically, progressive supranuclear palsy is characterized by accumulation of tau protein aggregates in the basal ganglia, brainstem, and cerebral cortex. The degenerative process involves dopaminergic, cholinergic, and gamma-aminobutyric acid (GABA)-ergic neurons.24
Gait and balance problems predominate early in progressive supranuclear palsy
The most commonly used diagnostic criteria are from the National Institute of Neurological Disorders and Stroke. The diagnosis of probable progressive supranuclear palsy requires vertical gaze palsy and falls or the tendency to fall within the first year of disease onset and exclusion of other causes.
The earliest symptom is usually gait and balance impairment.25 Falls (usually backward) and postural instability occur during the first year in 58% of patients.26 Instead of turning en bloc as in Parkinson disease, patients with progressive supranuclear palsy tend to pivot quickly. Patients may also have a coarse groaning voice and moaning. Insomnia has been reported, but rapid-eye-movement sleep behavior disorders are infrequent (unlike in Parkinson disease, multiple system atrophy, and Lewy body dementia).27
Apathy and extreme mood swings
Cognitive impairment is seen in 50% of patients in the early stage of progressive supranuclear palsy. It mostly involves the frontal lobe, including frontal behavioral disturbances (eg, apathy in 91% of patients26 or pseudobulbar affect and extreme emotional lability) and deficits in abstract thoughts or verbal fluency (to test this, patients are asked to say as many words as possible from a category in a given time). Ideomotor apraxia (inability to correctly imitate hand gestures and voluntarily pantomime tool use, such as pretending to brush hair) is rare, despite corticobasal degeneration.28
Vertical gaze palsy
The hallmark of progressive supranuclear palsy is vertical gaze palsy. Initially, this involves slowing of vertical saccades, followed by diminished vertical gaze and more characteristic downward gaze palsy. These findings may develop over 3 to 4 years. Vertical gaze palsy leads to spilling food and tripping while walking.
The gaze abnormality combined with rare blinking and facial dystonia form the classic facial expression of astonishment called “leonine facies.” The face is stiff and deeply furrowed, with a look of surprise.
Axial (especially neck) rigidity is more prominent than limb rigidity. Retrocollis (the head is drawn back) occurs in less than 25% of patients. Parkinsonian features such as bradykinesia affect nearly half of patients by the time of diagnosis.
Instead of the classic symptoms of progressive supranuclear palsy, about one-third of patients present with progressive supranuclear palsy-parkinsonism, which involves asymmetric parkinsonism that initially responds to levodopa.29
MRI shows ‘hummingbird sign’
Brain MRI shows atrophy of the brainstem, particularly the midbrain. Thinning of the superior part of the midbrain and dilation of the third ventricle (“hummingbird sign” on sagittal sections or “morning glory flower” on axial sections) support a diagnosis of progressive supranuclear palsy and differentiate it from Parkinson disease and other atypical parkinsonian disorders.30,31
Levodopa ineffective for supranuclear palsy
There is no treatment to slow progressive supranuclear palsy. Even in high doses, levodopa rarely alleviates parkinsonian features in a clinically meaningful way.26 Successful experimental biologic therapies have been studied in animal models.32 Davunetide is thought to help with neuronal integrity and cell survival through the stabilization of microtubules in preclinical studies, but it has not been used in clinical practice.33
CORTICOBASAL DEGENERATION
Corticobasal degeneration is a progressive, asymmetric movement disorder often manifesting initially with cognitive or behavioral impairment. It is associated with abnormality of the cytoskeleton protein tau. Onset is usually after age 60.
Asymmetric movement disorder with cognitive dysfunction
This diagnosis is clinical. Diagnostic criteria proposed in 2003 include the following core features34:
- Insidious onset and progressive course
- No identifiable cause
- Cortical dysfunction with at least one of the following: apraxia, alien limb phenomenon (one limb moves involuntarily with complex movements, eg, grabbing the other hand), cortical sensory loss, visual hemineglect, nonfluent aphasia
- Extrapyramidal dysfunction: focal rigidity unresponsive to levodopa, asymmetric dystonia.
An international consortium has developed more specific clinical research criteria for probable and possible corticobasal degeneration.35 In a series of 147 patients, the following clinical features were found: parkinsonism (100%), higher cortical dysfunction (93%), dyspraxia (82%), gait disorder (80%), unilateral limb dystonia (71%), tremor (55%), and dementia (25%).36
Behavioral problems commonly include depression; apathy, irritability, and agitation are also reported.37
Cognitive testing may reveal deficits in frontal-parietal cognitive domains including attention and concentration, executive function, verbal fluency, and visuospatial skills.38 Learning disabilities may be improved with verbal cueing (in contrast to Alzheimer disease). Patients may also have impaired graphesthesia (the ability to recognize writing on the skin only by the sensation of touch).39,40
Motor examination may reveal marked asymmetry. Hand, limb, speech, and gait apraxias are common. Gait is typically slow, with short steps and shuffling, and a wide-based or freezing gait. Arm swing may be absent on one side.
Asymmetric cortical atrophy
Early on, MRI may be normal. As the disease progresses, asymmetric cortical atrophy may be seen, especially in the posterior frontal and parietal lobes.
Levodopa ineffective in corticobasal degeneration
Corticobasal degeneration responds poorly to levodopa. Botulinum toxin has been used to help with dystonia and limb pain.
MULTIPLE SYSTEM ATROPHY
Multiple system atrophy is another atypical parkinsonian disorder, most often diagnosed in men over age 60. It is characterized by sporadic parkinsonism, cerebellar signs (involving balance and coordination), pyramidal tract dysfunction, and autonomic insufficiency in varying combinations. Two major subtypes are recognized, depending on whether the predominating presenting features are cerebellar signs or parkinsonism. In contrast to dementia with Lewy bodies, psychiatric symptoms are not a major feature, except possibly depression.41
Diagnosis requires a sporadic progressive disorder that has features of autonomic failure and poor response of parkinsonism or cerebellar ataxia to levodopa.42
Multiple system atrophy is usually not associated with dementia in the early stages, but patients develop deficits in learning, recognition, memory, and verbal fluency as the disease progresses.43 Rapid-eye-movement sleep behavior disorder has been reported in more than half of patients.44
A neurologic examination provides clues
Parkinsonian features are usually symmetric, in contrast to idiopathic Parkinson disease. These signs may include akinesia with rigidity, postural instability, hypokinetic speech, and tremor.
Cerebellar signs include nystagmus and dysarthria (speech disturbance), and gait and limb ataxia.
Pyramidal features include extensor plantar responses and hyperreflexia.
Autonomic dysfunction includes orthostatic hypotension, bladder and rectal atony, loss of sweating, urinary or fecal incontinence, and erectile dysfunction.
Electromyography may demonstrate decreased anal sphincter tone.
MRI shows atrophy of putamen and pons
Brain MRI may show atrophy of the putamen (hypointensity of the putamen with a hyperintense rim). Pons atrophy may also be present, revealing a “hot cross bun” sign in axial images. These combined findings have specificity above 90% but limited sensitivity. These signs are useful to distinguish multiple system atrophy from Parkinson dementia, but their absence does not exclude the diagnosis of multiple system atrophy.45,46
Multiple system atrophy typically responds poorly to levodopa
Levodopa may improve movement and rigidity, but many respond poorly to treatment or lose response after a few years. Fludrocortisone (Florinef) or vasoconstrictors such as midodrine (Orvaten, Proamatine) may help with orthostatic hypotension.47,48
PARKINSON DISEASE DEMENTIA
Dementia eventually develops in most patients with Parkinson disease. Older age and the akinetic rigid form of the disease are associated with higher risk. Diagnosis of idiopathic Parkinson disease before the development of dementia is essential for the diagnosis.
The Movement Disorder Society Task Force has developed new diagnostic criteria.49 Deficits must be present in at least two of the four core cognitive domains (attention, memory, executive, and visuospatial functions) and must be severe enough to affect daily functioning.
Behavioral symptoms such as affective changes, hallucinations, and apathy are common.
MRI shows characteristic brain atrophy in Parkinson disease dementia
MRI shows reduced gray matter volume in the frontal lobe in patients with Parkinson disease without dementia compared with controls. In Parkinson disease dementia, reduced volume extends to temporal, occipital, and subcortical areas. No significant volumetric differences have been observed in Parkinson dementia compared with dementia with Lewy bodies.50 A greater decrease of glucose metabolism has been found in the inferior parietal and occipital lobes in Parkinson disease dementia than in Parkinson disease without dementia.51
Rivastigmine effective for dementia
A Cochrane review supports the use of acetylcholinesterase inhibitors in patients with Parkinson disease dementia, with a positive impact on global assessment, cognitive function, behavioral disturbance, and activities of daily living rating scales.19 At this time, rivastigmine is the only FDA-approved cholinesterase inhibitor for treating Parkinson disease dementia. In clinical trials, memantine did not improve global clinical status or behavioral symptoms of dementia of Parkinson disease.51
FRONTOTEMPORAL DEMENTIA
Frontotemporal dementia frequently starts before age 65 and accounts for 20% to 50% of dementias in this age group.52 Recognition of the condition in older patients is also growing.53 Frontotemporal dementia encompasses a spectrum of dementias, including behavioral variant frontotemporal dementia, semantic dementia, and progressive nonfluent aphasia.54
Gradual onset of uncharacteristic behaviors
Accepted diagnostic criteria include core features of gradual onset, early decline in social and interpersonal conduct, early impairment of self-regulation, emotional blunting, and loss of insight. Many patients are diagnosed with psychiatric conditions. Changes reported by family and caregivers typically deviate substantially from the person’s usual behavior, such as impulsive and inappropriate behaviors or complete withdrawal and apathy.
Language sometimes affected in frontotemporal dementia
Language impairment may be present in some variants. Behavioral and language changes often accompany other forms of dementia (Alzheimer disease, vascular dementia, primary progressive aphasia), making diagnosis more challenging. Office-based testing often does not reveal any deficits, although the Frontal Behavioral Inventory may help.55 A referral to a clinical neuropsychologist may help identify and quantify cognitive impairments.
MRI shows frontotemporal lobes affected
Structural neuroimaging may not reveal abnormalities initially, but with progression, atrophy may be seen in the frontal and temporal lobes. Functional neuroimaging (positron emission tomography, brain SPECT, functional MRI) show hypometabolism in the same areas.
Treat symptoms
There are no specific FDA-approved therapies for frontotemporal dementia. Acetylcholinesterase inhibitors can help progressive nonfluent aphasia in some cases. Selective serotonin reuptake inhibitors may alleviate depressive symptoms, and low doses of atypical antipsychotic medications may help with impulsivity, disinhibition, and aggressive or disruptive behaviors.56
PRIMARY PROGRESSIVE APHASIA
Language impairment predominates
Primary progressive aphasia is a rare form of dementia in which symptoms typically develop around age 60. Pathology is varied. In a study of 60 patients with initial clinical symptoms of primary progressive aphasia, postmortem histology of brain tissue revealed various findings, including those consistent with Alzheimer pathology and motor neuron diseasetype inclusions.57
Patients typically present with expressive language problems as the primary deficit for the first 2 years of the disease, with preservation in other cognitive areas such as memory, visuospatial skills, and executive function.58 Office-based testing may overstate the severity of the dementia, given the dependence of performance on intact language.
It is important to distinguish primary progressive aphasia from other dementias that also affect language. In the frontal variant of frontotemporal dementia, the primary language problem is anomia (inability to name objects) or diminished speech output, which may be accompanied by behavioral problems. Semantic dementia affects word recognition as well as comprehension. In Alzheimer disease, language may be affected along with memory and other areas of cognitive function.
Imaging shows focal degeneration in the left hemisphere
Structural neuroimaging does not initially reveal any deficits, but later it may reveal atrophy in the frontal, perisylvian complex, and temporal areas of the left hemisphere, reflecting the focal nature of the degeneration.59 Functional neuroimaging (positron emission tomography, SPECT) may reveal hypometabolism or diminished blood flow in these areas prior to changes in structural neuroimaging.60
Other communication methods may help
There are no FDA-approved therapies for primary progressive aphasia. Off-label use of some agents (eg, selective serotonin reuptake inhibitors and small doses of antipsychotic medications) has been found useful in small trials.56 Patients may benefit from learning other forms of communication, such as using sign language, laminated cards with printed words or pictures, or artificial voice synthesizers, to express their needs.
NORMAL-PRESSURE HYDROCEPHALUS
Classic triad: Gait, cognition, incontinence
With the onset of symptoms in the sixth or seventh decade, normal-pressure hydrocephalus affects less than 1% of people age 65 and older. It represents up to 5% of dementias, although estimates are influenced by the varied criteria for diagnosis.61 It is characterized by the classic triad of gait impairment, cognitive impairment, and urinary frequency or incontinence.62
Symptoms progress over a period of years, with gait impairment often predominating. As this triad is common in the geriatric population, identifying other explanations is important. Gait impairment caused by spinal stenosis, peripheral neuropathy, or parkinsonism should be explored. Cognitive impairment could be due to depression, Alzheimer disease, or other forms of dementia. Urinary symptoms may be related to detrusor instability or an enlarged prostate.
Gait impairment initially manifests as slowing of gait, but progresses to difficulty with gait initiation. Gait tends to be wide-based (stance more than 1 foot wide).
Cognitive impairment is typically subcortical, manifested as slowed processing speed and impaired executive function. Recall and working memory may be impaired.
Enlarged ventricles seen on imaging in normal-pressure hydrocephalus
Structural neuroimaging reveals enlarged ventricles (Evan’s ratio > 0.358). This can be difficult to distinguish from ventriculomegaly due to cerebral atrophy; assessing the callosal angle on MRI may distinguish the two.63,64 Diagnosis of normal-pressure hydrocephalus can be confirmed using a cerebrospinal fluid infusion test to assess resistance of fluid to resorption.65
Treat with cerebrospinal fluid drainage
Specific tests should be performed to determine candidacy for surgery. These include a high-volume lumbar puncture (40 to 50 mL) or a trial of external lumbar drainage (10 mL per hour for 48 to 72 hours).65 Definitive treatment is surgical placement of a shunt to allow cerebrospinal fluid to drain into the atria or peritoneal cavity.
Surgery may improve gait, but cognitive symptoms often remain,66 and clinical decline may occur after the shunt is placed. Once gait dysfunction is resolved, other explanations for cognitive impairment or residual gait impairment should be considered. An underlying reason for progression of normal-pressure hydrocephalus symptoms after surgical intervention should be identified.67
RAPIDLY PROGRESSIVE DEMENTIAS
Rapidly progressive dementias are among the most challenging of dementing illnesses. They are characterized by a subacute course and an accelerated rate of decline, developing in less than 2 years. Evaluation should typically be more comprehensive than for other types of dementia. The main goal is to diagnose potentially treatable conditions, such as Hashimoto encephalopathy or paraneoplastic limbic encephalitis, and to distinguish these conditions from diseases with a very poor prognosis, such as Creutzfeldt-Jakob disease.
Creutzfeldt-Jakob disease
Creutzfeldt-Jakob disease is a fatal prion-related neurodegenerative illness. Sporadic disease is most common, but variant, familial, and iatrogenic types have been reported. The most common initial symptoms in sporadic disease are cognitive (39%), cerebellar (21%), behavioral (20%), constitutional (20%), sensory (11%), motor (9%), and visual (7%).68
Chronic neurodegenerative diseases can be misdiagnosed as Creutzfeldt-Jakob disease because of an atypical time course and multi-system neurologic findings.
The US Centers for Disease Control and Prevention has adopted criteria for diagnosing probable Creutzfeldt-Jakob disease (Table 3). Routine investigations should also not suggest an alternative diagnosis.69
Autoimmune diseases
Autoimmune conditions may present as a rapidly progressive dementia, including Hashimoto encephalopathy and antibody-mediated limbic encephalitis, either associated with cancer (paraneoplastic) or without cancer (nonparaneoplastic).
Paraneoplastic limbic encephalitis is a group of inflammatory conditions involving antibodies produced within the cerebrospinal fluid and serum resulting in neurologic symptoms. These antibodies react against proteins expressed mostly by a tumor somewhere else in the body.70
Hashimoto encephalitis is a subacute to chronic encephalopathy that may present as dementia with abnormally high levels of thyroid antibodies. The symptoms can vary from confusion to psychosis. There are two main presentations: one involves a relapsing-remitting course with stroke-like episodes (27% of patients) and the second consists of insidious onset of seizures (66% of patients).
Diagnosis involves testing for elevated anti-thyroid peroxidase and thyroglobulin antibodies. MRI findings are nonspecific. Hashimoto encephalitis responds to treatment with corticosteroids, plasmapheresis, or immunosuppressive therapy.71
- Plassman BL, Langa KM, Fisher GG, et al. Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology 2007; 29:125–132.
- Alzheimer’s Association. 2013 Alzheimer’s disease facts and figures. http://www.alz.org/downloads/facts_figures_2013.pdf. Accessed February 3, 2014.
- Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 2013; 80:1778–1783.
- Hou CE, Carlin D, Miller BL. Non-Alzheimer’s disease dementias: anatomic, clinical, and molecular correlates. Can J Psychiatry 2004; 49:164–171.
- Hachinski VC, Iliff LD, Zilhka E, et al. Cerebral blood flow in dementia. Arch Neurol 1975; 32:632–637.
- Fereshtehnejad SM, Religa D, Westman E, Aarsland D, Lökk J, Eriksdotter M. Demography, diagnostics, and medication in dementia with Lewy bodies and Parkinson’s disease with dementia: data from the Swedish Dementia Quality Registry (SveDem). Neuropsychiatr Dis Treat 2013; 9:927–935.
- Nomura T, Inoue Y, Takigawa H, Nakashima K. Comparison of REM sleep behaviour disorder variables between patients with progressive supranuclear palsy and those with Parkinson’s disease. Parkinsonism Relat Disord 2012; 18:394–396.
- Davis PH, Golbe LI, Duvoisin RC, Schoenberg BS. Risk factors for progrssive supranuclear palsy. Neurology 1988; 38:1546–1552.
- Tousi B, Schuele SU, Subramanian T. A 46-year-old woman with rigidity and frequent falls. Cleve Clin J Med 2005; 72:57–63.
- Cooper AD, Josephs KA. Photophobia, visual hallucinations, and REM sleep behavior disorder in progressive supranuclear palsy and corticobasal degeneration: a prospective study. Parkinsonism Relat Disord 2009; 15:59–61.
- Lyketsos CG, Lopez O, Jones B, Fitzpatrick AL, Breitner J, DeKosky S. Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: results from the cardiovascular health study0. JAMA 2002; 288:1475–1483.
- Knopman DS. Dementia and cerebrovascular disease. Mayo Clin Proc 2006; 81:223–230.
- Erkinjuntti T, Kurz A, Gauthier S, Bullock R, Lilienfeld S, Damaraju CV. Efficacy of galantamine in probable vascular dementia and Alzheimer’s disease combined with cerebrovascular disease: a randomised trial. Lancet 2002; 359:1283–1290.
- Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 2005; 293:596–608.
- Schneider LS, Dagerman K, Insel PS. Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebo-controlled trials. Am J Geriatr Psychiatry 2006; 14:191–210.
- McKeith IG, Dickson DW, Lowe J, et al; Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005; 65:1863–1872.
- Tartaglia MC, Rosen HJ, Miller BL. Neuroimaging in dementia. Neurotherapeutics 2011; 8:82–92.
- McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet 2000; 356:2031–2036.
- Rolinski M, Fox C, Maidment I, McShane R. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson’s disease dementia and cognitive impairment in Parkinson’s disease. Cochrane Database Syst Rev 2012; 3:CD006504.
- Emre M, Tsolaki M, Bonucelli U, et al; on behalf of the 11018 Study Investigators. Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010; 9:969–977.
- Kurlan R, Cummings J, Raman R, Thal L; Alzheimer’s Disease Cooperative Study Group. Quetiapine for agitation or psychosis in patients with dementia and parkinsonism. Neurology 2007; 68:1356–1363.
- Ballard C, Aarsland D, Francis P, Corbett A. Neuropsychiatric symptoms in patients with dementias associated with cortical Lewy bodies: pathophysiology, clinical features, and pharmacological management. Drugs Aging 2013; 30:603–611.
- Goetz CG, Leurgans S, Lang AE, Litvan I. Progression of gait, speech and swallowing deficits in progressive supranuclear palsy. Neurology 2003; 60:917–922.
- Kasashima S, Oda Y. Cholinergic neuronal loss in the basal forebrain and mesopontine tegmentum of progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol 2003; 105:117–124.
- Fahn S, Jankovic J, Hallett M, editors. Principles and Practice of Movement Disorders. 2nd ed. New York, NY: Elsevier/Saunders; 2011.
- Litvan I, Mangone CA, McKee A, et al. Natural history of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) and clinical predictors of survival: a clinicopathological study. J Neurol Neurosurg Psychiatry 1996; 60:615–620.
- Boeve BF, Silber MH, Parisi JE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology 2003; 61:40–45.
- Pharr V, Uttl B, Stark M, Litvan I, Fantie B, Grafman J. Comparison of apraxia in corticobasal degeneration and progressive supranuclear palsy. Neurology 2001; 56:957–963.
- Williams DR, Lees AJ. What features improve the accuracy of the clinical diagnosis of progressive supranuclear palsy-parkinsonism (PSP-P)? Mov Disord 2010; 25:357–362.
- Wenning GK, Litvan I, Tolosa E. Milestones in atypical and secondary parkinsonisms. Mov Disord 2011; 26:1083–1095.
- Gallucci M, Limbucci N, Catalucci A, Caulo M. Neurodegenerative diseases. Radiol Clin North Am 2008; 46:799–817.
- Stamelou M, de Silva R, Arias-Carrión O, et al. Rational therapeutic approaches to progressive supranuclear palsy. Brain 2010; 133:1578–1590.
- Gold M, Lorenzl S, Stewart AJ, Morimoto BH, Williams DR, Gozes I. Critical appraisal of the role of davunetide in the treatment of progressive supranuclear palsy. Neuropsychiatr Dis Treat 2012; 8:85–93.
- Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 2003; 54(suppl 5):S15–S19.
- Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013; 80:496–503.
- Kompoliti K, Goetz CG, Boeve BF, et al. Clinical presentation and pharmacological therapy in corticobasal degeneration. Arch Neurol 1998; 55:957–961.
- Litvan I, Cummings JL, Mega M. Neuropsychiatric features of corticobasal degeneration. J Neurol Neurosurg Psychiatry 1998; 65:717–721.
- Pillon B, Blin J, Vidailhet M, et al. The neuropsychological pattern of corticobasal degeneration: comparison with progressive supranuclear palsy and Alzheimer’s disease. Neurology 1995; 45:1477–1483.
- Tang-Wai DF, Josephs KA, Boeve BF, Petersen RC, Parisi JE, Dickson DW. Coexistent Lewy body disease in a case of “visual variant of Alzheimer’s disease.” J Neurol Neurosurg Psychiatry 2003; 74:389.
- Tang-Wai DF, Josephs KA, Boeve BF, Dickson DW, Parisi JE, Petersen RC. Pathologically confirmed corticobasal degeneration presenting with visuospatial dysfunction. Neurology 2003; 61:1134–1135.
- Goto K, Ueki A, Shimode H, Shinjo H, Miwa C, Morita Y. Depression in multiple system atrophy: a case report. Psychiatry Clin Neurosci 2000; 54:507–511.
- Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008; 71:670–676.
- Berent S, Giordani B, Gilman S, et al. Patterns of neuropsychological performance in multiple system atrophy compared to sporadic and hereditary olivopontocerebellar atrophy. Brain Cogn 2002; 50:194–206.
- Ghorayeb I, Yekhlef F, Chrysostome V, Balestre E, Bioulac B, Tison F. Sleep disorders and their determinants in multiple system atrophy. J Neurol Neurosurg Psychiatry 2002; 72:798–800.
- Schrag A, Kingsley D, Phatouros C, et al. Clinical usefulness of magnetic resonance imaging in multiple system atrophy. J Neurol Neurosurg Psychiatry 1998; 65:65–71.
- Massey LA, Micallef C, Paviour DC, et al. Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Mov Disord 2012; 27:1754–1762.
- Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA 1997; 277:1046–1051.
- Mathias CJ, Kimber JR. Postural hypotension: causes, clinical features, investigation, and management. Annu Rev Med 1999; 50:317–336.
- Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord 2007; 22:1689–1707.
- Burton EJ, McKeith IG, Burn DJ, Williams ED, O’Brien JT. Cerebral atrophy in Parkinson’s disease with and without dementia: a comparison with Alzheimer’s disease, dementia with Lewy bodies and controls. Brain 2004; 127:791–800.
- Piert M, Koeppe RA, Giordani B, Minoshima S, Kuhl DE. Determination of regional rate constants from dynamic FDG-PET studies in Parkinson’s disease. J Nucl Med 1996; 37:1115–1122.
- Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology 2002; 58:1615–1621.
- Baborie A, Griffiths TD, Jaros E, et al. Frontotemporal dementia in elderly individuals. Arch Neurol 2012; 69:1052–1960.
- Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998; 51:1546–1554.
- Kertesz A, Nadkarni N, Davidson W, Thomas AW. The Frontal Behavioral Inventory in the differential diagnosis of frontotemporal dementia. J Int Neuropsychol Soc 2000; 6:460–468.
- Mendez MF, Lauterbach EC, Sampson SM; ANPA Committee on Research. An evidence-based review of the psychopathology of frontotemporal dementia: a report of the ANPA Committee on Research. J Neuropsychiatry Clin Neurosci 2008; 20:130–149.
- Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain 2005; 128:1996–2005.
- Mesulam MM. Primary progressive aphasia—a language-based dementia. N Engl J Med 2003; 349:1535–1542.
- Turner RS, Kenyon LC, Trojanowski JQ, Gonatas N, Grossman M. Clinical, neuroimaging, and pathologic features of progressive nonfluent aphasia. Ann Neurol 1996; 39:166–173.
- Abe K, Ukita H, Yanagihara T. Imaging in primary progressive aphasia. Neuroradiology 1997; 39:556–559.
- Trenkwalder C, Schwarz J, Gebhard J, et al. Starnberg trial on epidemiology of parkinsonism and hypertension in the elderly. Prevalence of Parkinson’s disease and related disorders assessed by a door-to-door survey of inhabitants older than 65 years. Arch Neurol 1995; 52:1017–1022.
- Hakim S, Adams RD. The special clinical problem of symptomatic hydrocephalus with normal cerebrospinal fluid pressure. Observations on cerebrospinal fluid hydrodynamics. J Neurol Sci 1965; 2:307–327.
- Vanneste JA. Diagnosis and management of normal-pressure hydrocephalus. J Neurol 2000; 247:5–14.
- Ishii K, Kanda T, Harada A, et al. Clinical impact of the callosal angle in the diagnosis of idiopathic normal pressure hydrocephalus. Eur Radiol 2008; 18:2678–2683.
- Marmarou A, Bergsneider M, Klinge P, Relkin N, Black PM. The value of supplemental prognostic tests for the preoperative assessment of idiopathic normal-pressure hydrocephalus. Neurosurgery 2005; 57(suppl 3):S17–S28.
- Bergsneider M, Miller C, Vespa PM, Hu X. Surgical management of adult hydrocephalus. Neurosurgery 2008; 62(suppl 2):643–659.
- Malm J, Graff-Radford NR, Ishikawa M, et al. Influence of comorbidities in idiopathic normal pressure hydrocephalus—research and clinical care. A report of the ISHCSF task force on comorbidities in INPH. Fluids Barriers CNS 2013; 10:22.
- Rabinovici GD, Wang PN, Levin J, et al. First symptom in sporadic Creutzfeldt-Jakob disease. Neurology 2006; 66:286–287.
- Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 2009; 132:2659–2668.
- Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol 2008; 7:327–340.
- Chong JY, Rowland LP, Utiger RD. Hashimoto encephalopathy: syndrome or myth? Arch Neurol 2003; 60:164–171.
- Plassman BL, Langa KM, Fisher GG, et al. Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology 2007; 29:125–132.
- Alzheimer’s Association. 2013 Alzheimer’s disease facts and figures. http://www.alz.org/downloads/facts_figures_2013.pdf. Accessed February 3, 2014.
- Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 2013; 80:1778–1783.
- Hou CE, Carlin D, Miller BL. Non-Alzheimer’s disease dementias: anatomic, clinical, and molecular correlates. Can J Psychiatry 2004; 49:164–171.
- Hachinski VC, Iliff LD, Zilhka E, et al. Cerebral blood flow in dementia. Arch Neurol 1975; 32:632–637.
- Fereshtehnejad SM, Religa D, Westman E, Aarsland D, Lökk J, Eriksdotter M. Demography, diagnostics, and medication in dementia with Lewy bodies and Parkinson’s disease with dementia: data from the Swedish Dementia Quality Registry (SveDem). Neuropsychiatr Dis Treat 2013; 9:927–935.
- Nomura T, Inoue Y, Takigawa H, Nakashima K. Comparison of REM sleep behaviour disorder variables between patients with progressive supranuclear palsy and those with Parkinson’s disease. Parkinsonism Relat Disord 2012; 18:394–396.
- Davis PH, Golbe LI, Duvoisin RC, Schoenberg BS. Risk factors for progrssive supranuclear palsy. Neurology 1988; 38:1546–1552.
- Tousi B, Schuele SU, Subramanian T. A 46-year-old woman with rigidity and frequent falls. Cleve Clin J Med 2005; 72:57–63.
- Cooper AD, Josephs KA. Photophobia, visual hallucinations, and REM sleep behavior disorder in progressive supranuclear palsy and corticobasal degeneration: a prospective study. Parkinsonism Relat Disord 2009; 15:59–61.
- Lyketsos CG, Lopez O, Jones B, Fitzpatrick AL, Breitner J, DeKosky S. Prevalence of neuropsychiatric symptoms in dementia and mild cognitive impairment: results from the cardiovascular health study0. JAMA 2002; 288:1475–1483.
- Knopman DS. Dementia and cerebrovascular disease. Mayo Clin Proc 2006; 81:223–230.
- Erkinjuntti T, Kurz A, Gauthier S, Bullock R, Lilienfeld S, Damaraju CV. Efficacy of galantamine in probable vascular dementia and Alzheimer’s disease combined with cerebrovascular disease: a randomised trial. Lancet 2002; 359:1283–1290.
- Sink KM, Holden KF, Yaffe K. Pharmacological treatment of neuropsychiatric symptoms of dementia: a review of the evidence. JAMA 2005; 293:596–608.
- Schneider LS, Dagerman K, Insel PS. Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebo-controlled trials. Am J Geriatr Psychiatry 2006; 14:191–210.
- McKeith IG, Dickson DW, Lowe J, et al; Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005; 65:1863–1872.
- Tartaglia MC, Rosen HJ, Miller BL. Neuroimaging in dementia. Neurotherapeutics 2011; 8:82–92.
- McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet 2000; 356:2031–2036.
- Rolinski M, Fox C, Maidment I, McShane R. Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson’s disease dementia and cognitive impairment in Parkinson’s disease. Cochrane Database Syst Rev 2012; 3:CD006504.
- Emre M, Tsolaki M, Bonucelli U, et al; on behalf of the 11018 Study Investigators. Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010; 9:969–977.
- Kurlan R, Cummings J, Raman R, Thal L; Alzheimer’s Disease Cooperative Study Group. Quetiapine for agitation or psychosis in patients with dementia and parkinsonism. Neurology 2007; 68:1356–1363.
- Ballard C, Aarsland D, Francis P, Corbett A. Neuropsychiatric symptoms in patients with dementias associated with cortical Lewy bodies: pathophysiology, clinical features, and pharmacological management. Drugs Aging 2013; 30:603–611.
- Goetz CG, Leurgans S, Lang AE, Litvan I. Progression of gait, speech and swallowing deficits in progressive supranuclear palsy. Neurology 2003; 60:917–922.
- Kasashima S, Oda Y. Cholinergic neuronal loss in the basal forebrain and mesopontine tegmentum of progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol 2003; 105:117–124.
- Fahn S, Jankovic J, Hallett M, editors. Principles and Practice of Movement Disorders. 2nd ed. New York, NY: Elsevier/Saunders; 2011.
- Litvan I, Mangone CA, McKee A, et al. Natural history of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) and clinical predictors of survival: a clinicopathological study. J Neurol Neurosurg Psychiatry 1996; 60:615–620.
- Boeve BF, Silber MH, Parisi JE, et al. Synucleinopathy pathology and REM sleep behavior disorder plus dementia or parkinsonism. Neurology 2003; 61:40–45.
- Pharr V, Uttl B, Stark M, Litvan I, Fantie B, Grafman J. Comparison of apraxia in corticobasal degeneration and progressive supranuclear palsy. Neurology 2001; 56:957–963.
- Williams DR, Lees AJ. What features improve the accuracy of the clinical diagnosis of progressive supranuclear palsy-parkinsonism (PSP-P)? Mov Disord 2010; 25:357–362.
- Wenning GK, Litvan I, Tolosa E. Milestones in atypical and secondary parkinsonisms. Mov Disord 2011; 26:1083–1095.
- Gallucci M, Limbucci N, Catalucci A, Caulo M. Neurodegenerative diseases. Radiol Clin North Am 2008; 46:799–817.
- Stamelou M, de Silva R, Arias-Carrión O, et al. Rational therapeutic approaches to progressive supranuclear palsy. Brain 2010; 133:1578–1590.
- Gold M, Lorenzl S, Stewart AJ, Morimoto BH, Williams DR, Gozes I. Critical appraisal of the role of davunetide in the treatment of progressive supranuclear palsy. Neuropsychiatr Dis Treat 2012; 8:85–93.
- Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 2003; 54(suppl 5):S15–S19.
- Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013; 80:496–503.
- Kompoliti K, Goetz CG, Boeve BF, et al. Clinical presentation and pharmacological therapy in corticobasal degeneration. Arch Neurol 1998; 55:957–961.
- Litvan I, Cummings JL, Mega M. Neuropsychiatric features of corticobasal degeneration. J Neurol Neurosurg Psychiatry 1998; 65:717–721.
- Pillon B, Blin J, Vidailhet M, et al. The neuropsychological pattern of corticobasal degeneration: comparison with progressive supranuclear palsy and Alzheimer’s disease. Neurology 1995; 45:1477–1483.
- Tang-Wai DF, Josephs KA, Boeve BF, Petersen RC, Parisi JE, Dickson DW. Coexistent Lewy body disease in a case of “visual variant of Alzheimer’s disease.” J Neurol Neurosurg Psychiatry 2003; 74:389.
- Tang-Wai DF, Josephs KA, Boeve BF, Dickson DW, Parisi JE, Petersen RC. Pathologically confirmed corticobasal degeneration presenting with visuospatial dysfunction. Neurology 2003; 61:1134–1135.
- Goto K, Ueki A, Shimode H, Shinjo H, Miwa C, Morita Y. Depression in multiple system atrophy: a case report. Psychiatry Clin Neurosci 2000; 54:507–511.
- Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008; 71:670–676.
- Berent S, Giordani B, Gilman S, et al. Patterns of neuropsychological performance in multiple system atrophy compared to sporadic and hereditary olivopontocerebellar atrophy. Brain Cogn 2002; 50:194–206.
- Ghorayeb I, Yekhlef F, Chrysostome V, Balestre E, Bioulac B, Tison F. Sleep disorders and their determinants in multiple system atrophy. J Neurol Neurosurg Psychiatry 2002; 72:798–800.
- Schrag A, Kingsley D, Phatouros C, et al. Clinical usefulness of magnetic resonance imaging in multiple system atrophy. J Neurol Neurosurg Psychiatry 1998; 65:65–71.
- Massey LA, Micallef C, Paviour DC, et al. Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Mov Disord 2012; 27:1754–1762.
- Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA 1997; 277:1046–1051.
- Mathias CJ, Kimber JR. Postural hypotension: causes, clinical features, investigation, and management. Annu Rev Med 1999; 50:317–336.
- Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord 2007; 22:1689–1707.
- Burton EJ, McKeith IG, Burn DJ, Williams ED, O’Brien JT. Cerebral atrophy in Parkinson’s disease with and without dementia: a comparison with Alzheimer’s disease, dementia with Lewy bodies and controls. Brain 2004; 127:791–800.
- Piert M, Koeppe RA, Giordani B, Minoshima S, Kuhl DE. Determination of regional rate constants from dynamic FDG-PET studies in Parkinson’s disease. J Nucl Med 1996; 37:1115–1122.
- Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology 2002; 58:1615–1621.
- Baborie A, Griffiths TD, Jaros E, et al. Frontotemporal dementia in elderly individuals. Arch Neurol 2012; 69:1052–1960.
- Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998; 51:1546–1554.
- Kertesz A, Nadkarni N, Davidson W, Thomas AW. The Frontal Behavioral Inventory in the differential diagnosis of frontotemporal dementia. J Int Neuropsychol Soc 2000; 6:460–468.
- Mendez MF, Lauterbach EC, Sampson SM; ANPA Committee on Research. An evidence-based review of the psychopathology of frontotemporal dementia: a report of the ANPA Committee on Research. J Neuropsychiatry Clin Neurosci 2008; 20:130–149.
- Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain 2005; 128:1996–2005.
- Mesulam MM. Primary progressive aphasia—a language-based dementia. N Engl J Med 2003; 349:1535–1542.
- Turner RS, Kenyon LC, Trojanowski JQ, Gonatas N, Grossman M. Clinical, neuroimaging, and pathologic features of progressive nonfluent aphasia. Ann Neurol 1996; 39:166–173.
- Abe K, Ukita H, Yanagihara T. Imaging in primary progressive aphasia. Neuroradiology 1997; 39:556–559.
- Trenkwalder C, Schwarz J, Gebhard J, et al. Starnberg trial on epidemiology of parkinsonism and hypertension in the elderly. Prevalence of Parkinson’s disease and related disorders assessed by a door-to-door survey of inhabitants older than 65 years. Arch Neurol 1995; 52:1017–1022.
- Hakim S, Adams RD. The special clinical problem of symptomatic hydrocephalus with normal cerebrospinal fluid pressure. Observations on cerebrospinal fluid hydrodynamics. J Neurol Sci 1965; 2:307–327.
- Vanneste JA. Diagnosis and management of normal-pressure hydrocephalus. J Neurol 2000; 247:5–14.
- Ishii K, Kanda T, Harada A, et al. Clinical impact of the callosal angle in the diagnosis of idiopathic normal pressure hydrocephalus. Eur Radiol 2008; 18:2678–2683.
- Marmarou A, Bergsneider M, Klinge P, Relkin N, Black PM. The value of supplemental prognostic tests for the preoperative assessment of idiopathic normal-pressure hydrocephalus. Neurosurgery 2005; 57(suppl 3):S17–S28.
- Bergsneider M, Miller C, Vespa PM, Hu X. Surgical management of adult hydrocephalus. Neurosurgery 2008; 62(suppl 2):643–659.
- Malm J, Graff-Radford NR, Ishikawa M, et al. Influence of comorbidities in idiopathic normal pressure hydrocephalus—research and clinical care. A report of the ISHCSF task force on comorbidities in INPH. Fluids Barriers CNS 2013; 10:22.
- Rabinovici GD, Wang PN, Levin J, et al. First symptom in sporadic Creutzfeldt-Jakob disease. Neurology 2006; 66:286–287.
- Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 2009; 132:2659–2668.
- Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol 2008; 7:327–340.
- Chong JY, Rowland LP, Utiger RD. Hashimoto encephalopathy: syndrome or myth? Arch Neurol 2003; 60:164–171.
KEY POINTS
- Vascular dementia presents as a sudden, stepwise progression of cognitive deficits.
- Lewy body dementia often involves prominent visual hallucinations.
- Progressive supranuclear palsy starts with gait and balance problems caused by downward-gaze palsy.
- Many neurodegenerative conditions involve parkinsonism, but unlike Parkinson disease, they do not tend to respond well to levodopa, and dementia develops early.
- Corticobasal degeneration involves markedly asymmetric parkinsonism.
- Frontotemporal dementia involves dramatic behavior changes, including inappropriate impulsivity and complete apathy.
- Patients with rapidly progressive dementia should be evaluated for a treatable condition such as antibody-mediated encephalitis.
Movement disorder emergencies in the elderly: Recognizing and treating an often-iatrogenic problem
Although we tend to think of movement disorders as chronic conditions, some of them can present as true emergencies in which failure to diagnose the condition and treat it promptly can result in significant sickness or even death.
Many cases are iatrogenic, occurring in patients with Parkinson disease or those taking antipsychotic or antidepressant medications when their regimen is started or altered. Elderly patients are particularly at risk, as they take more drugs and have less physiologic reserve.
Movement disorder emergencies in elderly patients can be difficult to diagnose and treat, since many patients are taking more than one medication: polypharmacy raises the possibility of interactions, and different drugs can cause different movement disorder syndromes. Moreover, because so many patients are at risk—for example, more than 1 million people in the United States now have Parkinson disease, and the number is growing—it is important for physicians who take care of the elderly to be more informed about these disorders, especially the presenting symptoms.
SCOPE OF THIS ARTICLE

- Disorders presenting with rigidity or stiffness
- Disorders presenting with dystonia
- Disorders presenting with hyperkinetic movements
- Disorders presenting with psychiatric disturbances.
Of these, the scenarios most likely to require emergency evaluation in the elderly are acute hypokinetic and hyperkinetic syndromes and psychiatric presentations. This article discusses movement disorder emergencies in the elderly, focusing on the more common disorders with common presentations.
DISORDERS PRESENTING WITH RIGIDITY OR STIFFNESS
Serotonin syndrome

Chief among the offenders are the selective serotonin-reuptake inhibitors (SSRIs), either alone or in combination. This syndrome occurs in 14% to 16% of patients who overdose on SSRIs.1 Examples of combinations that can lead to serotonin syndrome are an SSRI plus any of the following:
- An anxiolytic such as buspirone (BuSpar; this combination is popular for the treatment of depression and anxiety)
- A tricyclic agent such as imipramine (Tofranil)
- A serotonin and norepinephrine reuptake inhibitor such as venlafaxine (Effexor).
In addition, antiparkinson drugs such as levodopa and selegiline (Eldepryl) enhance serotonin release.
Signs and symptoms. Serotonin syndrome is characterized by:
- Severe rigidity
- Dysautonomia
- Change in mental status.
Other clinical findings include fever, gastrointestinal disturbances, and motor restlessness. Clonus is the most important finding in establishing the diagnosis.2
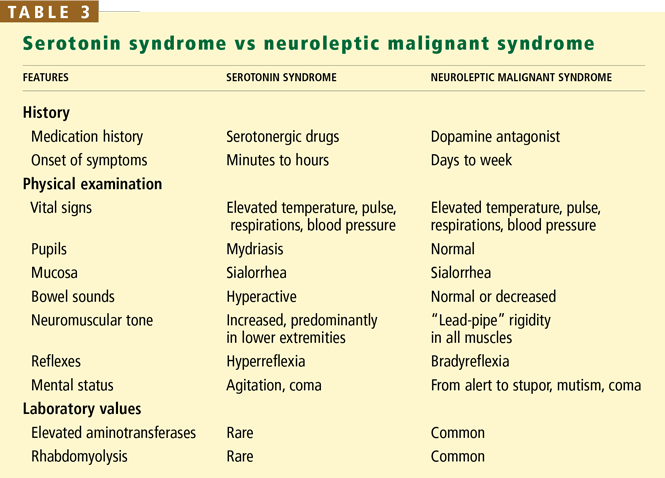
The syndrome may initially go unrecognized and can be mistaken for viral illness or anxiety.4 Manifestations range from mild to life-threatening; initially, it may present with akathisia and tremor. The symptoms progress rapidly over hours and can range from myoclonus, hyperreflexia, and seizures to severe forms of rhabdomyolysis, renal failure, and respiratory failure. The hyperreflexia and clonus seen in moderate cases may be considerably greater in the lower extremities than in the upper extremities.5
No laboratory test confirms the diagnosis, but tremor, clonus, or akathisia without additional extrapyramidal signs should lead to the diagnosis if the patient was taking a serotonergic medication.5 The onset of symptoms is usually rapid. The majority of patients present within 6 hours after initial use of the medication, an overdose, or a change in dosing.5
Treatment. The first steps are to stop the serotonergic medication and to hydrate and cool the patient to counteract the hyperpyrexic state. Benzodiazepine drugs are important in controlling agitation, regardless of its severity.5 Propranolol (Inderal) is not recommended, as it may cause hypotension and shock in patients with autonomic instability.5
Patients with moderate cases may additionally benefit from cyproheptadine (Periactin), an antihistamine that antagonizes serotonin. The initial dose is 4 to 8 mg orally, with a repeat dose after 2 hours.6 Whether to continue this treatment depends on the response after two doses.
If medications must be given parenterally, physicians can consider chlorpromazine (Thorazine) 50 to 100 mg intramuscularly.5
Vital signs should be monitored. In severe cases, intensive care may be required with immediate sedation, neuromuscular paralysis, and intubation.
In most cases, patients improve rapidly.
Comment. Serotonin syndrome can be avoided by educating physicians and by modifying prescribing practices.5 Avoiding multidrug regimens is critical to preventing serotonin syndrome. Computer-based ordering systems and personal digital assistants can help one avoid drug interactions.5
Neuroleptic malignant syndrome
This syndrome is an infrequent but potentially lethal complication associated with therapy with antipsychotic drugs such as haloperidol (Haldol) and lithium (Eskalith) and with other medications with dopamine type-2 receptor antagonist activity such as metoclopramide (Reglan) and prochlorperazine (Compazine). It has become rare since the introduction of atypical antipsychotics and now occurs in 0.2% of patients receiving atypical antipsychotics.7 Its pathogenesis is not fully understood.
This syndrome occurs mainly in young or middle-aged patients receiving doses of neuroleptics within the usual therapeutic range, but it also appears to occur in elderly patients who receive higher doses.8 Although most cases develop in the first 2 weeks of treatment, it can develop at any time during therapy.
Signs and symptoms. Four features characterize neuroleptic malignant syndrome9:
- Muscle rigidity—generalized (“lead-pipe”) muscular rigidity is accompanied by bradykinesia or akinesia.
- Autonomic dysregulation, with tachycardia, tachypnea, alterations in blood pressure, excessive sweating, and incontinence.
- Hyperthermia—fever can begin hours to days after initiating or increasing the dose of a dopamine antagonist.
- Altered sensorium, ranging from confusion to disorientation and coma.
Symptoms of neuroleptic malignant syndrome typically evolve over several days, in contrast to the rapid onset of the serotonin syndrome. Knowing the precipitating drug also helps distinguish the syndromes: dopamine antagonists produce bradykinesia, whereas serotonin agonists produce hyperkinesia.5
Laboratory abnormalities include elevated serum creatine kinase concentrations and white blood cell counts. Renal function should be assessed when renal failure and rhabdomyolysis are suspected.
Treatment involves stopping the causative medication, cooling the patient, and supporting vital functions.
In mild cases (eg, low-grade fever) benzodiazepines such as lorazepam (Ativan) can stabilize the condition. In moderate cases (eg, more significant rigidity), dopaminergic agonists such as bromocriptine (Parlodel) can be given, although there is no strong clinical evidence for their use. Bromocriptine is usually started at 2.5 mg three times a day and gradually increased in dose if tolerated.
In severe cases, muscle rigidity can be reduced with dantrolene (Dantrium), a muscle relaxant. Dantrolene is started at 1 mg/kg intravenously every 6 hours and gradually increased up to 10 mg/kg total per day.
Some patients remain rigid and febrile up to 4 weeks after the causative agent has been withdrawn. Therefore, these treatments can be continued for a few weeks. After the patient has recovered fully, if it is necessary to resume antipsychotic therapy, an atypical antipsychotic such as quetiapine (Seroquel) can be started after 2 weeks.8
Comment. Although uncommon, neuroleptic malignant syndrome is the most serious adverse effect of neuroleptic drugs, and it is potentially fatal. When neuroleptic malignant syndrome is suspected, treatment should be prompt, and the neuroleptic medication should be immediately stopped.
Parkinsonism-hyperpyrexia syndrome
Withdrawing or decreasing the dose of dopaminergic medications in patients with Parkinson disease can cause parkinsonism-hyperpyrexia syndrome, a condition that is similar to neuroleptic malignant syndrome. It can also arise after sudden withdrawal of amantadine (Symmetrel) or anticholinergics. In view of this concern, adjustments to antiparkinson drugs may need to be more gradual in some elderly patients.
Patients present with fever, rigidity, and autonomic instability and are at risk of aspiration pneumonia.
Treatment includes resuming dopaminergic therapy and giving supportive care.
Apomorphine (Apokyn), a dopaminergic agonist, was used in a 71-year-old female parkinsonian patient who developed parkinsonism-hyperpyrexia syndrome after abrupt reduction of chronic levodopa treatment.10 The symptoms resolved within 24 hours of the addition of apomorphine to her previous levodopa therapy. If the patient is taking apomorphine for the first time, the injections should be given in low doses, 0.2 mL subcutaneously. Apomorphine can induce vomiting, and if this occurs an antiemetic such as trimethobenzamide (Tigan) should be given before subsequent injections. In the elderly, caution is advised as apomorphine may cause severe orthostasis.
Methylprednisolone (Solu-Medrol) pulse therapy has been shown to shorten the duration of this syndrome in a randomized, controlled study.11
Akinetic syndrome after failure of deep brain stimulator
Deep brain stimulation involves surgical placement of a pacemaker with electrodes in specific areas of the brain. It is used to control Parkinson disease, tremor, and, less commonly, dystonia, and a number of other uses are under investigation. Continuous electrical stimulation of different nuclei in the brain has been shown to alleviate some symptoms of Parkinson disease (eg, rigidity) and to enable some patients to decrease the dose of their antiparkinson medications.
Several cases have been reported of sudden, unexpected reappearance of freezing, gait disturbance, or severe akinesia in Parkinson disease patients whose stimulators had been turned off inadvertently (eg, by a magnet in a dicating machine that was placed too close to the stimulator) and who presented to an emergency room.12
Treatment is easy if this diagnosis is considered. Checking the neurostimulator and switching it to “on” are all that is needed. Since patients and their caregivers are trained how to check and turn on the stimulator, the role of the geriatrician is simply to remind the caregiver of this possibility.
FDA warning. The US Food and Drug Administration has issued a warning against use of shortwave or microwave diathermy for patients with deep brain stimulation or other implanted leads (www.fda.gov/cdrh/safety/121902.html), stating: “There are three types of diathermy equipment used by physicians, dentists, physical therapists, chiropractors, sports therapists, and others: radio frequency (shortwave) diathermy, microwave diathermy, and ultrasound diathermy. Shortwave and microwave diathermy, in both heating and nonheating modes, can result in serious injury or death to patients with implanted devices with leads. This kind of interaction is not expected with ultrasound diathermy. Electrocautery devices are not included in this notification.” If a patient has an implanted deep brain stimulator, magnetic resonance imaging should be done only if absolutely needed and then only if the guidelines are followed.
DISORDERS PRESENTING WITH DYSTONIA
Acute dystonic reaction
Medications are a common cause of acute focal dystonia. The symptoms, which can be life-threatening, usually occur within 24 hours after taking the medication.12 The most common offenders are neuroleptic drugs and antiemetic drugs with dopamine-blocking activity (eg, metoclopramide), although in older patients, they are more likely to cause tardive dyskinesia and parkinsonism.13,14
Metoclopramide accounts for nearly one-third of all drug-induced movement disorders, and this adverse effect is a common reason for malpractice suits. The entire spectrum of drug-induced movement disorders, ranging from subtle to life-threatening, can ensue from its use; akathisia and dystonia are generally seen early in the course of metoclopramide-induced movement disorders, whereas tardive dyskinesia and parkinsonism seem to be more prevalent in long-term users.15
Treatment includes stopping the precipitating medication and reversing dystonia with anticholinergic medications such as benztropine (Cogentin). Anticholinergic therapy is given intravenously or intramuscularly followed by oral therapy for few days, as the acute dystonic reaction may recur after the effect of parenteral medication wears off.
Intravenous diphenhydramine (Benadryl), an antihistamine with additional anticholiner-gic effects, can abort dystonia in a few minutes.16
Laryngeal dystonia accompanied by multiple system atrophy
Multiple system atrophy, a Parkinson-plus syndrome, is characterized by parkinsonism (mostly with poor response to levodopa) and early onset of dysfunction of the autonomic nervous system, urinary tract, cerebellum, and corticospinal tract (hyperreflexia).17
In the course of the disease, about one-third of patients develop respiratory stridor due to abnormal movements of the vocal cords.18 Nocturnal stridor portends a poor prognosis,19 with an increased risk of sudden death. Geriatricians should be aware of these symptoms, as these patients may seek care because of hoarseness or difficulty swallowing.
Treatment. Laryngeal dystonia can be improved with continuous positive airway pressure. In some cases, tracheostomy may be needed.19
Sudden withdrawal of baclofen
Baclofen (Lioresal), a treatment for spasticity and dystonia, is delivered via a pump through a catheter into an intrathecal space. The pump needs to be refilled every 3 to 6 months. Sudden discontinuation of medication caused by a dislodged catheter tip or forgetting to refill the pump provokes withdrawal symptoms. Patients with this life-threatening syndrome can present with rigidity, fever, change in mental status, and worsening dystonic symptoms.
Treatment involves high doses of baclofen (up to 120 mg/day in divided doses).6
DISORDERS PRESENTING WITH HYPERKINETIC MOVEMENTS
Chorea, ballism (ballismus), and athetosis constitute a range of involuntary, hyperkinetic movement disorders. Chorea consists of involuntary, continuous, sudden, brief, unsustained, irregular movements that flow from one part of the body to another. Hemiballism presents as forceful flinging movements of the limbs or high-amplitude chorea that affects one side of the body.
Acute hemichorea and hemiballism
Acute hemichorea and hemiballism commonly result from infarction or hemorrhage of the basal ganglia.20 Computed tomography and especially magnetic resonance imaging can show the lesions in patients with ballism. Stroke-induced ballism is usually self-limited and resolves after a few weeks. Acute hemiballism generally evolves to hemichorea or hemiathetosis in a few days, which requires only protective measures.
Treatment. Mild cases do not need treatment but severe cases call for medical therapy. Antidopaminergics are the drugs of choice. A dopamine depletor such as reserpine (Serpasil) 0.1 mg once or twice daily or dopamine receptor blockers such as neuroleptics are considered.16 The combination of a benzodiazepine plus an antipsychotic such as olanzapine (Zyprexa) has been suggested.6
Severe parkinsonian dyskinesia
Dyskinesia is common in Parkinson disease, and patients may present to an emergency room with severe levodopa-induced dyskinesia. Dyskinesia can be exhausting if prolonged and severe. Elevated levels of creatine kinase raise the concern of rhabdomyolysis. In rare cases, the patient develops respiratory dyskinesia when respiratory muscles such as those in the diaphragm become involved.21
The risk of levodopa-induced dyskinesia increases with disease severity and higher levodopa doses. Using a dopamine agonist as initial therapy delays the onset of levodopa-induced dyskinesia in early Parkinson disease. However, Factor and Molho,21 in a case series, reported that adding dopamine agonists to the regimen was a precipitating factor; another was infection.
Treatment. A reasonable approach to treating peak-dose dyskinesia is to lower the doses of dopaminergic medications.
A mild sedative such as lorazepam, alprazolam (Xanax, Niravam), or clonazepam (Klonopin) may reduce the severity of dyski-nesia.21 These drugs are particularly useful if the dyskinesia is worse at night, and they can be used in the emergency department while waiting for the effect of the dopaminergic medications to wear off.
Amantadine ameliorates levodopa-induced peak-dose dyskinesia without worsening parkinsonian symptoms in some patients.22
Drug-induced myoclonus
Myoclonus is sudden, jerky, brief involuntary movement of the face, limbs, or trunk. Unlike tics, myoclonus cannot be controlled by the patient.
Myoclonus has various pathophysiologic mechanisms. Most myoclonic emergencies are epileptic myoclonic seizures, which are beyond the scope of this article. Often, myoclonus is caused by opiate overdose or withdrawal. It can also be a side effect of SSRIs, tricyclic anti-depressants, lithium, amantadine, and rarely, antibiotics such as imipenem (Primaxin).23
Treatment. Opiate-induced myoclonus may respond to naloxone (Narcan), whereas opiate withdrawal responds to benzodi-azepines.6
Acute akathisia
Acute akathisia occurs in susceptible patients after exposure to dopamine receptor blockers or dopamine depletors. It is characterized by subjective restless feelings accompanied by objective restless movements. The course is usually self-limited after the causative medication is discontinued.
Treatment. Symptomatic treatment may be needed in most cases for several days. Anticholinergics are effective. Additionally, vitamin B6, mianserine, propranolol, and mirtazapine (Remeron) in a low dose (15 mg/day) have been shown to be effective16,24,25
DISORDERS WITH PSYCHIATRIC PRESENTATIONS
Hallucinations and psychosis in Parkinson disease
Neuropsychiatric or behavioral complications of Parkinson disease include hallucinations, dementia, depression, psychosis, and sleep disorders.21,26 Psychosis is the leading reason for nursing home placement in advanced cases.27 Psychosis can present as hallucinations or a paranoid delusional state in association with clear sensorium.28 However, hallucinations accounted for only 3% of emergency admissions to the hospital for Parkinson disease patients in one series.29
Risk factors for hallucinations in parkinsonian patients include dementia, long-term therapy with dopaminergic drugs, long duration of disease, advanced age, anticholinergic drugs, and sleep disorders. Severe cognitive impairment or dementia is a major and independent predictive factor for visual hallucinations.30
Most hallucinations are visual; auditory, tactile, and olfactory hallucinations are rare.30
Treatment initially should be the same as in any patient with delirium. The systemic disorders that can aggravate or cause hallucinations such as electrolyte abnormalities, urinary or respiratory infection, and systemic illness should be ruled out.
The next step is to reduce or discontinue the adjunctive drugs that have the least antiparkinsonian effect and the greatest potential of inducing hallucination or psychosis. Examples of such medications include histamine-2 antagonists (eg, cimetidine [Tagamet], amantadine, selegeline, and anti-cholinergics). Selegeline can be discontinued abruptly because it has a long duration of action in the brain, but amantadine and anti-cholinergics should be tapered. Dopamine agonists can be discontinued. Levodopa can be reduced until the side effects begin to subside without significant worsening of motor symptoms.
If all the above adjustments fail, an antipsychotic medication can be considered.26 Clozapine (Clozaril) has the best result and is nearly free of extrapyramidal side effects but can cause agranulocytosis, which requires frequent blood counts. The Parkinson Study Group suggested that clozapine, at daily doses of 50 mg or less, is safe and significantly improves drug-induced psychosis without worsening parkinsonism.31 Clozapine may be impractical for elderly patients due to its side effect profile.
Quetiapine is a good alternative to cloza-pine and is less likely to worsen parkinsonian symptoms than other atypical antipsy-chotics.32 Olanzapine and risperidone (Risperdal) are reported to worsen parkinsonian symptoms.33 Not enough data have been published about the efficacy of the newer medications such as ziprasidone (Geodon) and aripiprazole (Abilify) to advocate their routine clinical use.
Rivastigmine (Exelon) was reported to improve hallucinations, sleep disturbance, and caregiver distress in addition to enhancing cognitive performance in advanced Parkinson disease in a small study.34 Burn and colleagues35 reported that rivastigmine was beneficial in patients with dementia associated with Parkinson disease, with or without hallucinations. Efficacy measures were cognitive scales, activities of daily living, behavioral symptoms, and executive and attentional functions. The differences in these measures between rivastigmine and placebo recipients tended to be larger in patients with visual hallucinations than in those without hallucinations. The study was not designed to assess the effect of treatment on psychosis or hallucination.
WHEN PATIENTS WITH MOVEMENT DISORDERS NEED SURGERY
Some of these syndromes can be prevented, especially in patients who are known to have movement disorders and are undergoing surgery.
One problem is stopping oral dopaminergic drugs before the operation. Parkinson disease patients on dopaminergic drugs can develop parkinsonism-hyperpyrexia syndrome or akinetic crisis if the drug is stopped suddenly. Restarting dopaminergic therapy and supportive measures are the main treatments. Patients who have Parkinson disease should receive their usual dose of levodopa, dopamine agonist, or amantadine up until the time of surgery and then again as soon as they awaken in the recovery room.36 That goal can be achieved more easily now that these drugs come in transdermal patches and long-acting formulas.37 Droperidol (Inapsine) and metoclopramide worsen parkinsonism and should be avoided.
Myoclonus is the most common movement disorder seen in the postoperative period. In fact, myoclonic shivering is common as patients awaken from general anesthesia.36 The anesthetic agents etomidate (Amidate) and enflurane (Ethrane) and the opioids fentanyl (Actiq, Duralgesic, Sublimaze) and meperidine (Demerol) can cause myoclonus.38
Occasionally, a patient in the recovery room suddenly develops a neurologic deficit that is inconsistent with the history and physical findings. Psychogenic movement disorders should be considered in the differential diagnosis. Reassurance and occasionally psychiatric intervention are required in these cases.36
IN THE ELDERLY, GO EASY
Polypharmacy is a huge issue in the elderly. Some of the principles in prescribing medications in the elderly can be helpful in preventing movement disorder emergencies:
- Assess the current regimen, including over-the-counter drugs, before prescribing a new drug.
- Begin with a low dose and increase as necessary. “Start low, go slow.”
- Consider the possibility that any new symptoms can be a drug side effect or due to withdrawal of a drug.
- Discuss with the patient or caregiver what kind of side effect to expect and advise him or her to report serious ones.
- Isbister GK, Bowe SJ, Dawson A, Whyte IM. Relative toxicity of selective serotonin reuptake inhibitors (SSRIs) in overdose. J Toxicol Clin Toxicol 2004; 42:277–285.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96:635–642.
- Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000; 79:201–209.
- LoCurto MJ. The serotonin syndrome. Emerg Med Clin North Am 1997; 15( 3):665–675.
- Boyer E, Shannon S. The serotonin syndrome. N Engl J Med 2005; 352:1112–1120.
- Kipps CM, Fung VS, Grattan-Smith P, de Moore GM, Morris JG. Movement disorder emergencies. Mov Disord 2005; 20:322–334.
- Shalev A, Munitz H. The neuroleptic malignant syndrome: agent and host interaction. Acta Psychiatr Scand 1986; 73:337–347.
- Rosebush PI, Stewart TD, Gelenberg AJ. Twenty neuroleptic rechallenges after neuroleptic malignant syndrome in 15 patients. J Clin Psychiatry 1989; 50:295–298.
- Adityanjee , Singh S, Singh G, Ong S. Spectrum concept of neuroleptic malignant syndrome. Br J Psychiatry 1988; 153:107–111.
- Bonuccelli U, Piccini P, Corsini GU, Muratorio A. Apomorphine in malignant syndrome due to levodopa withdrawal. Ital J Neurol Sci 1992; 13:169–170.
- Sato Y, Asoh T, Metoki N, et al. Efficacy of methylprednisolone pulse therapy on neuroleptic malignant syndrome in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2004; 74:574–576.
- Hariz MI, Johansson F. Hardware failure in parkinsonian patients with chronic subthalamic nucleus stimulation is a medical emergency. Mov Disord 2001; 16:166–168.
- Pollera CF, Cognetli F, Nardi M, Mozza D. Sudden death after acute dystonic reaction to high-dose metoclopramide. Lancet 1984; 2:460–461.
- Bateman DN, Rawlins MD, Simpson JM. Extrapyramidal reactions with metoclopramide. Br Med J 1985; 291:930–932.
- Pasricha PJ, Pehlivanov N, Sugumar A, Jankovic J. Drug insight: from disturbed motility to disordered movement—a review of the clinical benefits and medicolegal risks of metoclopramide. Nat Clin Pract Gastroenterol Hepatol 2006; 3:138–148.
- Hu S, Frucht S. Emergency treatment of movement disorders. Curr Treat Options Neurol 2007; 9:103–114.
- Tousi B, Schuele SU, Subramanian T. A 46-year-old woman with rigidity and frequent falls. Cleve Clin J Med 2005; 72:57–63.
- Merlo IM, Occhini A, Pacchetti C, Alfonsi E. Not paralysis, but dystonia causes stridor in multiple system atrophy. Neurology 2002; 58:649–652.
- Silber MH, Levine S. Stridor and death in multiple system atrophy. Mov Disord 2000; 15:699–704.
- Bhatia KP, Marsden CD. The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain 1994; 117:859–876.
- Factor SA, Molho ES. Emergency department presentations of patients with Parkinson’s disease. Am J Emerg Med 2000; 18:209–215.
- Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as a treatment for dyskinesia and motor fluctuations in Parkinson’s disease. Neurology 1998; 50:1323–1326.
- Frucht S, Eidelberg D. Imipenem-induced myoclonus. Mov Disord 1997; 12:621–622.
- Miodownik C, Lerner V, Statsenko N, et al. Vitamin B6 versus mianserin and placebo in acute neuroleptic-induced akathisia: a randomized, double-blind, controlled study. Clin Neuropharmacol 2006; 29:68–72.
- Poyurovsky M, Pashinian A, Weizman R, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry 2006; 59:1071–1077.
- Tousi B, Subramanian T. Hallucinations in Parkinson’s disease: approach and management. Clin Geriatr 2004: 12:19–24.
- Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology 1993; 43:2227–2229.
- Factor SA, Molho ES, Podskalny GD, Brown D. Parkinson’s disease: drug-induced psychiatric states. Adv Neurol 1995; 65:115–138.
- Woodford H, Walker R. Emergency hospital admissions in idiopathic Parkinson’s disease. Mov Disord 2005; 20:1104–1108.
- Tousi B, Frankel M. Olfactory and visual hallucinations in Parkinson’s disease. Parkinsonism Relat Disord 2004; 10:253–254.
- The Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson’s disease. N Engl J Med 1999; 340:757–763.
- Merims D, Balas M, Pertez C, Shabtai H, Giladi N. Rater-blinded, prospective comparison: quetiapine versus clozapine for Parkinson’s disease psychosis. Clin Neuropharmacol 2006; 29:331–337.
- Goetz CG, Blasucci LM, Leurgans S, Pappert EJ. Olanzapine and clozapine: comparative effects on motor function in hallucinating PD patients. Neurology 2000; 55:789–794.
- Reading PJ, Luce AK, McKeith IG. Rivastigmine in the treatment of parkinsonian psychosis and cognitive impairment: preliminary findings from an open trial. Mov Disord 2001; 16:1171–1174.
- Burn D, Emre M, McKeith I, et al. Effects of rivastigmine in patients with and without visual hallucinations in dementia associated with Parkinson’s disease. Mov Disord 2006; 21:1899–1907.
- Frucht SJ. Movement disorder emergencies in the perioperative period. Neurol Clin 2004; 22:379–387.
- Korczyn AD, Reichmann H, Boroojerdi B, et al. Rotigotin trans-dermal system for perioperative administration. J Neural Transm 2007; 114:219–221.
- Gordon MF. Toxin and drug-induced myoclonus. Adv Neurol 2002; 89:49–76.
Although we tend to think of movement disorders as chronic conditions, some of them can present as true emergencies in which failure to diagnose the condition and treat it promptly can result in significant sickness or even death.
Many cases are iatrogenic, occurring in patients with Parkinson disease or those taking antipsychotic or antidepressant medications when their regimen is started or altered. Elderly patients are particularly at risk, as they take more drugs and have less physiologic reserve.
Movement disorder emergencies in elderly patients can be difficult to diagnose and treat, since many patients are taking more than one medication: polypharmacy raises the possibility of interactions, and different drugs can cause different movement disorder syndromes. Moreover, because so many patients are at risk—for example, more than 1 million people in the United States now have Parkinson disease, and the number is growing—it is important for physicians who take care of the elderly to be more informed about these disorders, especially the presenting symptoms.
SCOPE OF THIS ARTICLE
- Disorders presenting with rigidity or stiffness
- Disorders presenting with dystonia
- Disorders presenting with hyperkinetic movements
- Disorders presenting with psychiatric disturbances.
Of these, the scenarios most likely to require emergency evaluation in the elderly are acute hypokinetic and hyperkinetic syndromes and psychiatric presentations. This article discusses movement disorder emergencies in the elderly, focusing on the more common disorders with common presentations.
DISORDERS PRESENTING WITH RIGIDITY OR STIFFNESS
Serotonin syndrome
Chief among the offenders are the selective serotonin-reuptake inhibitors (SSRIs), either alone or in combination. This syndrome occurs in 14% to 16% of patients who overdose on SSRIs.1 Examples of combinations that can lead to serotonin syndrome are an SSRI plus any of the following:
- An anxiolytic such as buspirone (BuSpar; this combination is popular for the treatment of depression and anxiety)
- A tricyclic agent such as imipramine (Tofranil)
- A serotonin and norepinephrine reuptake inhibitor such as venlafaxine (Effexor).
In addition, antiparkinson drugs such as levodopa and selegiline (Eldepryl) enhance serotonin release.
Signs and symptoms. Serotonin syndrome is characterized by:
- Severe rigidity
- Dysautonomia
- Change in mental status.
Other clinical findings include fever, gastrointestinal disturbances, and motor restlessness. Clonus is the most important finding in establishing the diagnosis.2
The syndrome may initially go unrecognized and can be mistaken for viral illness or anxiety.4 Manifestations range from mild to life-threatening; initially, it may present with akathisia and tremor. The symptoms progress rapidly over hours and can range from myoclonus, hyperreflexia, and seizures to severe forms of rhabdomyolysis, renal failure, and respiratory failure. The hyperreflexia and clonus seen in moderate cases may be considerably greater in the lower extremities than in the upper extremities.5
No laboratory test confirms the diagnosis, but tremor, clonus, or akathisia without additional extrapyramidal signs should lead to the diagnosis if the patient was taking a serotonergic medication.5 The onset of symptoms is usually rapid. The majority of patients present within 6 hours after initial use of the medication, an overdose, or a change in dosing.5
Treatment. The first steps are to stop the serotonergic medication and to hydrate and cool the patient to counteract the hyperpyrexic state. Benzodiazepine drugs are important in controlling agitation, regardless of its severity.5 Propranolol (Inderal) is not recommended, as it may cause hypotension and shock in patients with autonomic instability.5
Patients with moderate cases may additionally benefit from cyproheptadine (Periactin), an antihistamine that antagonizes serotonin. The initial dose is 4 to 8 mg orally, with a repeat dose after 2 hours.6 Whether to continue this treatment depends on the response after two doses.
If medications must be given parenterally, physicians can consider chlorpromazine (Thorazine) 50 to 100 mg intramuscularly.5
Vital signs should be monitored. In severe cases, intensive care may be required with immediate sedation, neuromuscular paralysis, and intubation.
In most cases, patients improve rapidly.
Comment. Serotonin syndrome can be avoided by educating physicians and by modifying prescribing practices.5 Avoiding multidrug regimens is critical to preventing serotonin syndrome. Computer-based ordering systems and personal digital assistants can help one avoid drug interactions.5
Neuroleptic malignant syndrome
This syndrome is an infrequent but potentially lethal complication associated with therapy with antipsychotic drugs such as haloperidol (Haldol) and lithium (Eskalith) and with other medications with dopamine type-2 receptor antagonist activity such as metoclopramide (Reglan) and prochlorperazine (Compazine). It has become rare since the introduction of atypical antipsychotics and now occurs in 0.2% of patients receiving atypical antipsychotics.7 Its pathogenesis is not fully understood.
This syndrome occurs mainly in young or middle-aged patients receiving doses of neuroleptics within the usual therapeutic range, but it also appears to occur in elderly patients who receive higher doses.8 Although most cases develop in the first 2 weeks of treatment, it can develop at any time during therapy.
Signs and symptoms. Four features characterize neuroleptic malignant syndrome9:
- Muscle rigidity—generalized (“lead-pipe”) muscular rigidity is accompanied by bradykinesia or akinesia.
- Autonomic dysregulation, with tachycardia, tachypnea, alterations in blood pressure, excessive sweating, and incontinence.
- Hyperthermia—fever can begin hours to days after initiating or increasing the dose of a dopamine antagonist.
- Altered sensorium, ranging from confusion to disorientation and coma.
Symptoms of neuroleptic malignant syndrome typically evolve over several days, in contrast to the rapid onset of the serotonin syndrome. Knowing the precipitating drug also helps distinguish the syndromes: dopamine antagonists produce bradykinesia, whereas serotonin agonists produce hyperkinesia.5
Laboratory abnormalities include elevated serum creatine kinase concentrations and white blood cell counts. Renal function should be assessed when renal failure and rhabdomyolysis are suspected.
Treatment involves stopping the causative medication, cooling the patient, and supporting vital functions.
In mild cases (eg, low-grade fever) benzodiazepines such as lorazepam (Ativan) can stabilize the condition. In moderate cases (eg, more significant rigidity), dopaminergic agonists such as bromocriptine (Parlodel) can be given, although there is no strong clinical evidence for their use. Bromocriptine is usually started at 2.5 mg three times a day and gradually increased in dose if tolerated.
In severe cases, muscle rigidity can be reduced with dantrolene (Dantrium), a muscle relaxant. Dantrolene is started at 1 mg/kg intravenously every 6 hours and gradually increased up to 10 mg/kg total per day.
Some patients remain rigid and febrile up to 4 weeks after the causative agent has been withdrawn. Therefore, these treatments can be continued for a few weeks. After the patient has recovered fully, if it is necessary to resume antipsychotic therapy, an atypical antipsychotic such as quetiapine (Seroquel) can be started after 2 weeks.8
Comment. Although uncommon, neuroleptic malignant syndrome is the most serious adverse effect of neuroleptic drugs, and it is potentially fatal. When neuroleptic malignant syndrome is suspected, treatment should be prompt, and the neuroleptic medication should be immediately stopped.
Parkinsonism-hyperpyrexia syndrome
Withdrawing or decreasing the dose of dopaminergic medications in patients with Parkinson disease can cause parkinsonism-hyperpyrexia syndrome, a condition that is similar to neuroleptic malignant syndrome. It can also arise after sudden withdrawal of amantadine (Symmetrel) or anticholinergics. In view of this concern, adjustments to antiparkinson drugs may need to be more gradual in some elderly patients.
Patients present with fever, rigidity, and autonomic instability and are at risk of aspiration pneumonia.
Treatment includes resuming dopaminergic therapy and giving supportive care.
Apomorphine (Apokyn), a dopaminergic agonist, was used in a 71-year-old female parkinsonian patient who developed parkinsonism-hyperpyrexia syndrome after abrupt reduction of chronic levodopa treatment.10 The symptoms resolved within 24 hours of the addition of apomorphine to her previous levodopa therapy. If the patient is taking apomorphine for the first time, the injections should be given in low doses, 0.2 mL subcutaneously. Apomorphine can induce vomiting, and if this occurs an antiemetic such as trimethobenzamide (Tigan) should be given before subsequent injections. In the elderly, caution is advised as apomorphine may cause severe orthostasis.
Methylprednisolone (Solu-Medrol) pulse therapy has been shown to shorten the duration of this syndrome in a randomized, controlled study.11
Akinetic syndrome after failure of deep brain stimulator
Deep brain stimulation involves surgical placement of a pacemaker with electrodes in specific areas of the brain. It is used to control Parkinson disease, tremor, and, less commonly, dystonia, and a number of other uses are under investigation. Continuous electrical stimulation of different nuclei in the brain has been shown to alleviate some symptoms of Parkinson disease (eg, rigidity) and to enable some patients to decrease the dose of their antiparkinson medications.
Several cases have been reported of sudden, unexpected reappearance of freezing, gait disturbance, or severe akinesia in Parkinson disease patients whose stimulators had been turned off inadvertently (eg, by a magnet in a dicating machine that was placed too close to the stimulator) and who presented to an emergency room.12
Treatment is easy if this diagnosis is considered. Checking the neurostimulator and switching it to “on” are all that is needed. Since patients and their caregivers are trained how to check and turn on the stimulator, the role of the geriatrician is simply to remind the caregiver of this possibility.
FDA warning. The US Food and Drug Administration has issued a warning against use of shortwave or microwave diathermy for patients with deep brain stimulation or other implanted leads (www.fda.gov/cdrh/safety/121902.html), stating: “There are three types of diathermy equipment used by physicians, dentists, physical therapists, chiropractors, sports therapists, and others: radio frequency (shortwave) diathermy, microwave diathermy, and ultrasound diathermy. Shortwave and microwave diathermy, in both heating and nonheating modes, can result in serious injury or death to patients with implanted devices with leads. This kind of interaction is not expected with ultrasound diathermy. Electrocautery devices are not included in this notification.” If a patient has an implanted deep brain stimulator, magnetic resonance imaging should be done only if absolutely needed and then only if the guidelines are followed.
DISORDERS PRESENTING WITH DYSTONIA
Acute dystonic reaction
Medications are a common cause of acute focal dystonia. The symptoms, which can be life-threatening, usually occur within 24 hours after taking the medication.12 The most common offenders are neuroleptic drugs and antiemetic drugs with dopamine-blocking activity (eg, metoclopramide), although in older patients, they are more likely to cause tardive dyskinesia and parkinsonism.13,14
Metoclopramide accounts for nearly one-third of all drug-induced movement disorders, and this adverse effect is a common reason for malpractice suits. The entire spectrum of drug-induced movement disorders, ranging from subtle to life-threatening, can ensue from its use; akathisia and dystonia are generally seen early in the course of metoclopramide-induced movement disorders, whereas tardive dyskinesia and parkinsonism seem to be more prevalent in long-term users.15
Treatment includes stopping the precipitating medication and reversing dystonia with anticholinergic medications such as benztropine (Cogentin). Anticholinergic therapy is given intravenously or intramuscularly followed by oral therapy for few days, as the acute dystonic reaction may recur after the effect of parenteral medication wears off.
Intravenous diphenhydramine (Benadryl), an antihistamine with additional anticholiner-gic effects, can abort dystonia in a few minutes.16
Laryngeal dystonia accompanied by multiple system atrophy
Multiple system atrophy, a Parkinson-plus syndrome, is characterized by parkinsonism (mostly with poor response to levodopa) and early onset of dysfunction of the autonomic nervous system, urinary tract, cerebellum, and corticospinal tract (hyperreflexia).17
In the course of the disease, about one-third of patients develop respiratory stridor due to abnormal movements of the vocal cords.18 Nocturnal stridor portends a poor prognosis,19 with an increased risk of sudden death. Geriatricians should be aware of these symptoms, as these patients may seek care because of hoarseness or difficulty swallowing.
Treatment. Laryngeal dystonia can be improved with continuous positive airway pressure. In some cases, tracheostomy may be needed.19
Sudden withdrawal of baclofen
Baclofen (Lioresal), a treatment for spasticity and dystonia, is delivered via a pump through a catheter into an intrathecal space. The pump needs to be refilled every 3 to 6 months. Sudden discontinuation of medication caused by a dislodged catheter tip or forgetting to refill the pump provokes withdrawal symptoms. Patients with this life-threatening syndrome can present with rigidity, fever, change in mental status, and worsening dystonic symptoms.
Treatment involves high doses of baclofen (up to 120 mg/day in divided doses).6
DISORDERS PRESENTING WITH HYPERKINETIC MOVEMENTS
Chorea, ballism (ballismus), and athetosis constitute a range of involuntary, hyperkinetic movement disorders. Chorea consists of involuntary, continuous, sudden, brief, unsustained, irregular movements that flow from one part of the body to another. Hemiballism presents as forceful flinging movements of the limbs or high-amplitude chorea that affects one side of the body.
Acute hemichorea and hemiballism
Acute hemichorea and hemiballism commonly result from infarction or hemorrhage of the basal ganglia.20 Computed tomography and especially magnetic resonance imaging can show the lesions in patients with ballism. Stroke-induced ballism is usually self-limited and resolves after a few weeks. Acute hemiballism generally evolves to hemichorea or hemiathetosis in a few days, which requires only protective measures.
Treatment. Mild cases do not need treatment but severe cases call for medical therapy. Antidopaminergics are the drugs of choice. A dopamine depletor such as reserpine (Serpasil) 0.1 mg once or twice daily or dopamine receptor blockers such as neuroleptics are considered.16 The combination of a benzodiazepine plus an antipsychotic such as olanzapine (Zyprexa) has been suggested.6
Severe parkinsonian dyskinesia
Dyskinesia is common in Parkinson disease, and patients may present to an emergency room with severe levodopa-induced dyskinesia. Dyskinesia can be exhausting if prolonged and severe. Elevated levels of creatine kinase raise the concern of rhabdomyolysis. In rare cases, the patient develops respiratory dyskinesia when respiratory muscles such as those in the diaphragm become involved.21
The risk of levodopa-induced dyskinesia increases with disease severity and higher levodopa doses. Using a dopamine agonist as initial therapy delays the onset of levodopa-induced dyskinesia in early Parkinson disease. However, Factor and Molho,21 in a case series, reported that adding dopamine agonists to the regimen was a precipitating factor; another was infection.
Treatment. A reasonable approach to treating peak-dose dyskinesia is to lower the doses of dopaminergic medications.
A mild sedative such as lorazepam, alprazolam (Xanax, Niravam), or clonazepam (Klonopin) may reduce the severity of dyski-nesia.21 These drugs are particularly useful if the dyskinesia is worse at night, and they can be used in the emergency department while waiting for the effect of the dopaminergic medications to wear off.
Amantadine ameliorates levodopa-induced peak-dose dyskinesia without worsening parkinsonian symptoms in some patients.22
Drug-induced myoclonus
Myoclonus is sudden, jerky, brief involuntary movement of the face, limbs, or trunk. Unlike tics, myoclonus cannot be controlled by the patient.
Myoclonus has various pathophysiologic mechanisms. Most myoclonic emergencies are epileptic myoclonic seizures, which are beyond the scope of this article. Often, myoclonus is caused by opiate overdose or withdrawal. It can also be a side effect of SSRIs, tricyclic anti-depressants, lithium, amantadine, and rarely, antibiotics such as imipenem (Primaxin).23
Treatment. Opiate-induced myoclonus may respond to naloxone (Narcan), whereas opiate withdrawal responds to benzodi-azepines.6
Acute akathisia
Acute akathisia occurs in susceptible patients after exposure to dopamine receptor blockers or dopamine depletors. It is characterized by subjective restless feelings accompanied by objective restless movements. The course is usually self-limited after the causative medication is discontinued.
Treatment. Symptomatic treatment may be needed in most cases for several days. Anticholinergics are effective. Additionally, vitamin B6, mianserine, propranolol, and mirtazapine (Remeron) in a low dose (15 mg/day) have been shown to be effective16,24,25
DISORDERS WITH PSYCHIATRIC PRESENTATIONS
Hallucinations and psychosis in Parkinson disease
Neuropsychiatric or behavioral complications of Parkinson disease include hallucinations, dementia, depression, psychosis, and sleep disorders.21,26 Psychosis is the leading reason for nursing home placement in advanced cases.27 Psychosis can present as hallucinations or a paranoid delusional state in association with clear sensorium.28 However, hallucinations accounted for only 3% of emergency admissions to the hospital for Parkinson disease patients in one series.29
Risk factors for hallucinations in parkinsonian patients include dementia, long-term therapy with dopaminergic drugs, long duration of disease, advanced age, anticholinergic drugs, and sleep disorders. Severe cognitive impairment or dementia is a major and independent predictive factor for visual hallucinations.30
Most hallucinations are visual; auditory, tactile, and olfactory hallucinations are rare.30
Treatment initially should be the same as in any patient with delirium. The systemic disorders that can aggravate or cause hallucinations such as electrolyte abnormalities, urinary or respiratory infection, and systemic illness should be ruled out.
The next step is to reduce or discontinue the adjunctive drugs that have the least antiparkinsonian effect and the greatest potential of inducing hallucination or psychosis. Examples of such medications include histamine-2 antagonists (eg, cimetidine [Tagamet], amantadine, selegeline, and anti-cholinergics). Selegeline can be discontinued abruptly because it has a long duration of action in the brain, but amantadine and anti-cholinergics should be tapered. Dopamine agonists can be discontinued. Levodopa can be reduced until the side effects begin to subside without significant worsening of motor symptoms.
If all the above adjustments fail, an antipsychotic medication can be considered.26 Clozapine (Clozaril) has the best result and is nearly free of extrapyramidal side effects but can cause agranulocytosis, which requires frequent blood counts. The Parkinson Study Group suggested that clozapine, at daily doses of 50 mg or less, is safe and significantly improves drug-induced psychosis without worsening parkinsonism.31 Clozapine may be impractical for elderly patients due to its side effect profile.
Quetiapine is a good alternative to cloza-pine and is less likely to worsen parkinsonian symptoms than other atypical antipsy-chotics.32 Olanzapine and risperidone (Risperdal) are reported to worsen parkinsonian symptoms.33 Not enough data have been published about the efficacy of the newer medications such as ziprasidone (Geodon) and aripiprazole (Abilify) to advocate their routine clinical use.
Rivastigmine (Exelon) was reported to improve hallucinations, sleep disturbance, and caregiver distress in addition to enhancing cognitive performance in advanced Parkinson disease in a small study.34 Burn and colleagues35 reported that rivastigmine was beneficial in patients with dementia associated with Parkinson disease, with or without hallucinations. Efficacy measures were cognitive scales, activities of daily living, behavioral symptoms, and executive and attentional functions. The differences in these measures between rivastigmine and placebo recipients tended to be larger in patients with visual hallucinations than in those without hallucinations. The study was not designed to assess the effect of treatment on psychosis or hallucination.
WHEN PATIENTS WITH MOVEMENT DISORDERS NEED SURGERY
Some of these syndromes can be prevented, especially in patients who are known to have movement disorders and are undergoing surgery.
One problem is stopping oral dopaminergic drugs before the operation. Parkinson disease patients on dopaminergic drugs can develop parkinsonism-hyperpyrexia syndrome or akinetic crisis if the drug is stopped suddenly. Restarting dopaminergic therapy and supportive measures are the main treatments. Patients who have Parkinson disease should receive their usual dose of levodopa, dopamine agonist, or amantadine up until the time of surgery and then again as soon as they awaken in the recovery room.36 That goal can be achieved more easily now that these drugs come in transdermal patches and long-acting formulas.37 Droperidol (Inapsine) and metoclopramide worsen parkinsonism and should be avoided.
Myoclonus is the most common movement disorder seen in the postoperative period. In fact, myoclonic shivering is common as patients awaken from general anesthesia.36 The anesthetic agents etomidate (Amidate) and enflurane (Ethrane) and the opioids fentanyl (Actiq, Duralgesic, Sublimaze) and meperidine (Demerol) can cause myoclonus.38
Occasionally, a patient in the recovery room suddenly develops a neurologic deficit that is inconsistent with the history and physical findings. Psychogenic movement disorders should be considered in the differential diagnosis. Reassurance and occasionally psychiatric intervention are required in these cases.36
IN THE ELDERLY, GO EASY
Polypharmacy is a huge issue in the elderly. Some of the principles in prescribing medications in the elderly can be helpful in preventing movement disorder emergencies:
- Assess the current regimen, including over-the-counter drugs, before prescribing a new drug.
- Begin with a low dose and increase as necessary. “Start low, go slow.”
- Consider the possibility that any new symptoms can be a drug side effect or due to withdrawal of a drug.
- Discuss with the patient or caregiver what kind of side effect to expect and advise him or her to report serious ones.
Although we tend to think of movement disorders as chronic conditions, some of them can present as true emergencies in which failure to diagnose the condition and treat it promptly can result in significant sickness or even death.
Many cases are iatrogenic, occurring in patients with Parkinson disease or those taking antipsychotic or antidepressant medications when their regimen is started or altered. Elderly patients are particularly at risk, as they take more drugs and have less physiologic reserve.
Movement disorder emergencies in elderly patients can be difficult to diagnose and treat, since many patients are taking more than one medication: polypharmacy raises the possibility of interactions, and different drugs can cause different movement disorder syndromes. Moreover, because so many patients are at risk—for example, more than 1 million people in the United States now have Parkinson disease, and the number is growing—it is important for physicians who take care of the elderly to be more informed about these disorders, especially the presenting symptoms.
SCOPE OF THIS ARTICLE
- Disorders presenting with rigidity or stiffness
- Disorders presenting with dystonia
- Disorders presenting with hyperkinetic movements
- Disorders presenting with psychiatric disturbances.
Of these, the scenarios most likely to require emergency evaluation in the elderly are acute hypokinetic and hyperkinetic syndromes and psychiatric presentations. This article discusses movement disorder emergencies in the elderly, focusing on the more common disorders with common presentations.
DISORDERS PRESENTING WITH RIGIDITY OR STIFFNESS
Serotonin syndrome
Chief among the offenders are the selective serotonin-reuptake inhibitors (SSRIs), either alone or in combination. This syndrome occurs in 14% to 16% of patients who overdose on SSRIs.1 Examples of combinations that can lead to serotonin syndrome are an SSRI plus any of the following:
- An anxiolytic such as buspirone (BuSpar; this combination is popular for the treatment of depression and anxiety)
- A tricyclic agent such as imipramine (Tofranil)
- A serotonin and norepinephrine reuptake inhibitor such as venlafaxine (Effexor).
In addition, antiparkinson drugs such as levodopa and selegiline (Eldepryl) enhance serotonin release.
Signs and symptoms. Serotonin syndrome is characterized by:
- Severe rigidity
- Dysautonomia
- Change in mental status.
Other clinical findings include fever, gastrointestinal disturbances, and motor restlessness. Clonus is the most important finding in establishing the diagnosis.2
The syndrome may initially go unrecognized and can be mistaken for viral illness or anxiety.4 Manifestations range from mild to life-threatening; initially, it may present with akathisia and tremor. The symptoms progress rapidly over hours and can range from myoclonus, hyperreflexia, and seizures to severe forms of rhabdomyolysis, renal failure, and respiratory failure. The hyperreflexia and clonus seen in moderate cases may be considerably greater in the lower extremities than in the upper extremities.5
No laboratory test confirms the diagnosis, but tremor, clonus, or akathisia without additional extrapyramidal signs should lead to the diagnosis if the patient was taking a serotonergic medication.5 The onset of symptoms is usually rapid. The majority of patients present within 6 hours after initial use of the medication, an overdose, or a change in dosing.5
Treatment. The first steps are to stop the serotonergic medication and to hydrate and cool the patient to counteract the hyperpyrexic state. Benzodiazepine drugs are important in controlling agitation, regardless of its severity.5 Propranolol (Inderal) is not recommended, as it may cause hypotension and shock in patients with autonomic instability.5
Patients with moderate cases may additionally benefit from cyproheptadine (Periactin), an antihistamine that antagonizes serotonin. The initial dose is 4 to 8 mg orally, with a repeat dose after 2 hours.6 Whether to continue this treatment depends on the response after two doses.
If medications must be given parenterally, physicians can consider chlorpromazine (Thorazine) 50 to 100 mg intramuscularly.5
Vital signs should be monitored. In severe cases, intensive care may be required with immediate sedation, neuromuscular paralysis, and intubation.
In most cases, patients improve rapidly.
Comment. Serotonin syndrome can be avoided by educating physicians and by modifying prescribing practices.5 Avoiding multidrug regimens is critical to preventing serotonin syndrome. Computer-based ordering systems and personal digital assistants can help one avoid drug interactions.5
Neuroleptic malignant syndrome
This syndrome is an infrequent but potentially lethal complication associated with therapy with antipsychotic drugs such as haloperidol (Haldol) and lithium (Eskalith) and with other medications with dopamine type-2 receptor antagonist activity such as metoclopramide (Reglan) and prochlorperazine (Compazine). It has become rare since the introduction of atypical antipsychotics and now occurs in 0.2% of patients receiving atypical antipsychotics.7 Its pathogenesis is not fully understood.
This syndrome occurs mainly in young or middle-aged patients receiving doses of neuroleptics within the usual therapeutic range, but it also appears to occur in elderly patients who receive higher doses.8 Although most cases develop in the first 2 weeks of treatment, it can develop at any time during therapy.
Signs and symptoms. Four features characterize neuroleptic malignant syndrome9:
- Muscle rigidity—generalized (“lead-pipe”) muscular rigidity is accompanied by bradykinesia or akinesia.
- Autonomic dysregulation, with tachycardia, tachypnea, alterations in blood pressure, excessive sweating, and incontinence.
- Hyperthermia—fever can begin hours to days after initiating or increasing the dose of a dopamine antagonist.
- Altered sensorium, ranging from confusion to disorientation and coma.
Symptoms of neuroleptic malignant syndrome typically evolve over several days, in contrast to the rapid onset of the serotonin syndrome. Knowing the precipitating drug also helps distinguish the syndromes: dopamine antagonists produce bradykinesia, whereas serotonin agonists produce hyperkinesia.5
Laboratory abnormalities include elevated serum creatine kinase concentrations and white blood cell counts. Renal function should be assessed when renal failure and rhabdomyolysis are suspected.
Treatment involves stopping the causative medication, cooling the patient, and supporting vital functions.
In mild cases (eg, low-grade fever) benzodiazepines such as lorazepam (Ativan) can stabilize the condition. In moderate cases (eg, more significant rigidity), dopaminergic agonists such as bromocriptine (Parlodel) can be given, although there is no strong clinical evidence for their use. Bromocriptine is usually started at 2.5 mg three times a day and gradually increased in dose if tolerated.
In severe cases, muscle rigidity can be reduced with dantrolene (Dantrium), a muscle relaxant. Dantrolene is started at 1 mg/kg intravenously every 6 hours and gradually increased up to 10 mg/kg total per day.
Some patients remain rigid and febrile up to 4 weeks after the causative agent has been withdrawn. Therefore, these treatments can be continued for a few weeks. After the patient has recovered fully, if it is necessary to resume antipsychotic therapy, an atypical antipsychotic such as quetiapine (Seroquel) can be started after 2 weeks.8
Comment. Although uncommon, neuroleptic malignant syndrome is the most serious adverse effect of neuroleptic drugs, and it is potentially fatal. When neuroleptic malignant syndrome is suspected, treatment should be prompt, and the neuroleptic medication should be immediately stopped.
Parkinsonism-hyperpyrexia syndrome
Withdrawing or decreasing the dose of dopaminergic medications in patients with Parkinson disease can cause parkinsonism-hyperpyrexia syndrome, a condition that is similar to neuroleptic malignant syndrome. It can also arise after sudden withdrawal of amantadine (Symmetrel) or anticholinergics. In view of this concern, adjustments to antiparkinson drugs may need to be more gradual in some elderly patients.
Patients present with fever, rigidity, and autonomic instability and are at risk of aspiration pneumonia.
Treatment includes resuming dopaminergic therapy and giving supportive care.
Apomorphine (Apokyn), a dopaminergic agonist, was used in a 71-year-old female parkinsonian patient who developed parkinsonism-hyperpyrexia syndrome after abrupt reduction of chronic levodopa treatment.10 The symptoms resolved within 24 hours of the addition of apomorphine to her previous levodopa therapy. If the patient is taking apomorphine for the first time, the injections should be given in low doses, 0.2 mL subcutaneously. Apomorphine can induce vomiting, and if this occurs an antiemetic such as trimethobenzamide (Tigan) should be given before subsequent injections. In the elderly, caution is advised as apomorphine may cause severe orthostasis.
Methylprednisolone (Solu-Medrol) pulse therapy has been shown to shorten the duration of this syndrome in a randomized, controlled study.11
Akinetic syndrome after failure of deep brain stimulator
Deep brain stimulation involves surgical placement of a pacemaker with electrodes in specific areas of the brain. It is used to control Parkinson disease, tremor, and, less commonly, dystonia, and a number of other uses are under investigation. Continuous electrical stimulation of different nuclei in the brain has been shown to alleviate some symptoms of Parkinson disease (eg, rigidity) and to enable some patients to decrease the dose of their antiparkinson medications.
Several cases have been reported of sudden, unexpected reappearance of freezing, gait disturbance, or severe akinesia in Parkinson disease patients whose stimulators had been turned off inadvertently (eg, by a magnet in a dicating machine that was placed too close to the stimulator) and who presented to an emergency room.12
Treatment is easy if this diagnosis is considered. Checking the neurostimulator and switching it to “on” are all that is needed. Since patients and their caregivers are trained how to check and turn on the stimulator, the role of the geriatrician is simply to remind the caregiver of this possibility.
FDA warning. The US Food and Drug Administration has issued a warning against use of shortwave or microwave diathermy for patients with deep brain stimulation or other implanted leads (www.fda.gov/cdrh/safety/121902.html), stating: “There are three types of diathermy equipment used by physicians, dentists, physical therapists, chiropractors, sports therapists, and others: radio frequency (shortwave) diathermy, microwave diathermy, and ultrasound diathermy. Shortwave and microwave diathermy, in both heating and nonheating modes, can result in serious injury or death to patients with implanted devices with leads. This kind of interaction is not expected with ultrasound diathermy. Electrocautery devices are not included in this notification.” If a patient has an implanted deep brain stimulator, magnetic resonance imaging should be done only if absolutely needed and then only if the guidelines are followed.
DISORDERS PRESENTING WITH DYSTONIA
Acute dystonic reaction
Medications are a common cause of acute focal dystonia. The symptoms, which can be life-threatening, usually occur within 24 hours after taking the medication.12 The most common offenders are neuroleptic drugs and antiemetic drugs with dopamine-blocking activity (eg, metoclopramide), although in older patients, they are more likely to cause tardive dyskinesia and parkinsonism.13,14
Metoclopramide accounts for nearly one-third of all drug-induced movement disorders, and this adverse effect is a common reason for malpractice suits. The entire spectrum of drug-induced movement disorders, ranging from subtle to life-threatening, can ensue from its use; akathisia and dystonia are generally seen early in the course of metoclopramide-induced movement disorders, whereas tardive dyskinesia and parkinsonism seem to be more prevalent in long-term users.15
Treatment includes stopping the precipitating medication and reversing dystonia with anticholinergic medications such as benztropine (Cogentin). Anticholinergic therapy is given intravenously or intramuscularly followed by oral therapy for few days, as the acute dystonic reaction may recur after the effect of parenteral medication wears off.
Intravenous diphenhydramine (Benadryl), an antihistamine with additional anticholiner-gic effects, can abort dystonia in a few minutes.16
Laryngeal dystonia accompanied by multiple system atrophy
Multiple system atrophy, a Parkinson-plus syndrome, is characterized by parkinsonism (mostly with poor response to levodopa) and early onset of dysfunction of the autonomic nervous system, urinary tract, cerebellum, and corticospinal tract (hyperreflexia).17
In the course of the disease, about one-third of patients develop respiratory stridor due to abnormal movements of the vocal cords.18 Nocturnal stridor portends a poor prognosis,19 with an increased risk of sudden death. Geriatricians should be aware of these symptoms, as these patients may seek care because of hoarseness or difficulty swallowing.
Treatment. Laryngeal dystonia can be improved with continuous positive airway pressure. In some cases, tracheostomy may be needed.19
Sudden withdrawal of baclofen
Baclofen (Lioresal), a treatment for spasticity and dystonia, is delivered via a pump through a catheter into an intrathecal space. The pump needs to be refilled every 3 to 6 months. Sudden discontinuation of medication caused by a dislodged catheter tip or forgetting to refill the pump provokes withdrawal symptoms. Patients with this life-threatening syndrome can present with rigidity, fever, change in mental status, and worsening dystonic symptoms.
Treatment involves high doses of baclofen (up to 120 mg/day in divided doses).6
DISORDERS PRESENTING WITH HYPERKINETIC MOVEMENTS
Chorea, ballism (ballismus), and athetosis constitute a range of involuntary, hyperkinetic movement disorders. Chorea consists of involuntary, continuous, sudden, brief, unsustained, irregular movements that flow from one part of the body to another. Hemiballism presents as forceful flinging movements of the limbs or high-amplitude chorea that affects one side of the body.
Acute hemichorea and hemiballism
Acute hemichorea and hemiballism commonly result from infarction or hemorrhage of the basal ganglia.20 Computed tomography and especially magnetic resonance imaging can show the lesions in patients with ballism. Stroke-induced ballism is usually self-limited and resolves after a few weeks. Acute hemiballism generally evolves to hemichorea or hemiathetosis in a few days, which requires only protective measures.
Treatment. Mild cases do not need treatment but severe cases call for medical therapy. Antidopaminergics are the drugs of choice. A dopamine depletor such as reserpine (Serpasil) 0.1 mg once or twice daily or dopamine receptor blockers such as neuroleptics are considered.16 The combination of a benzodiazepine plus an antipsychotic such as olanzapine (Zyprexa) has been suggested.6
Severe parkinsonian dyskinesia
Dyskinesia is common in Parkinson disease, and patients may present to an emergency room with severe levodopa-induced dyskinesia. Dyskinesia can be exhausting if prolonged and severe. Elevated levels of creatine kinase raise the concern of rhabdomyolysis. In rare cases, the patient develops respiratory dyskinesia when respiratory muscles such as those in the diaphragm become involved.21
The risk of levodopa-induced dyskinesia increases with disease severity and higher levodopa doses. Using a dopamine agonist as initial therapy delays the onset of levodopa-induced dyskinesia in early Parkinson disease. However, Factor and Molho,21 in a case series, reported that adding dopamine agonists to the regimen was a precipitating factor; another was infection.
Treatment. A reasonable approach to treating peak-dose dyskinesia is to lower the doses of dopaminergic medications.
A mild sedative such as lorazepam, alprazolam (Xanax, Niravam), or clonazepam (Klonopin) may reduce the severity of dyski-nesia.21 These drugs are particularly useful if the dyskinesia is worse at night, and they can be used in the emergency department while waiting for the effect of the dopaminergic medications to wear off.
Amantadine ameliorates levodopa-induced peak-dose dyskinesia without worsening parkinsonian symptoms in some patients.22
Drug-induced myoclonus
Myoclonus is sudden, jerky, brief involuntary movement of the face, limbs, or trunk. Unlike tics, myoclonus cannot be controlled by the patient.
Myoclonus has various pathophysiologic mechanisms. Most myoclonic emergencies are epileptic myoclonic seizures, which are beyond the scope of this article. Often, myoclonus is caused by opiate overdose or withdrawal. It can also be a side effect of SSRIs, tricyclic anti-depressants, lithium, amantadine, and rarely, antibiotics such as imipenem (Primaxin).23
Treatment. Opiate-induced myoclonus may respond to naloxone (Narcan), whereas opiate withdrawal responds to benzodi-azepines.6
Acute akathisia
Acute akathisia occurs in susceptible patients after exposure to dopamine receptor blockers or dopamine depletors. It is characterized by subjective restless feelings accompanied by objective restless movements. The course is usually self-limited after the causative medication is discontinued.
Treatment. Symptomatic treatment may be needed in most cases for several days. Anticholinergics are effective. Additionally, vitamin B6, mianserine, propranolol, and mirtazapine (Remeron) in a low dose (15 mg/day) have been shown to be effective16,24,25
DISORDERS WITH PSYCHIATRIC PRESENTATIONS
Hallucinations and psychosis in Parkinson disease
Neuropsychiatric or behavioral complications of Parkinson disease include hallucinations, dementia, depression, psychosis, and sleep disorders.21,26 Psychosis is the leading reason for nursing home placement in advanced cases.27 Psychosis can present as hallucinations or a paranoid delusional state in association with clear sensorium.28 However, hallucinations accounted for only 3% of emergency admissions to the hospital for Parkinson disease patients in one series.29
Risk factors for hallucinations in parkinsonian patients include dementia, long-term therapy with dopaminergic drugs, long duration of disease, advanced age, anticholinergic drugs, and sleep disorders. Severe cognitive impairment or dementia is a major and independent predictive factor for visual hallucinations.30
Most hallucinations are visual; auditory, tactile, and olfactory hallucinations are rare.30
Treatment initially should be the same as in any patient with delirium. The systemic disorders that can aggravate or cause hallucinations such as electrolyte abnormalities, urinary or respiratory infection, and systemic illness should be ruled out.
The next step is to reduce or discontinue the adjunctive drugs that have the least antiparkinsonian effect and the greatest potential of inducing hallucination or psychosis. Examples of such medications include histamine-2 antagonists (eg, cimetidine [Tagamet], amantadine, selegeline, and anti-cholinergics). Selegeline can be discontinued abruptly because it has a long duration of action in the brain, but amantadine and anti-cholinergics should be tapered. Dopamine agonists can be discontinued. Levodopa can be reduced until the side effects begin to subside without significant worsening of motor symptoms.
If all the above adjustments fail, an antipsychotic medication can be considered.26 Clozapine (Clozaril) has the best result and is nearly free of extrapyramidal side effects but can cause agranulocytosis, which requires frequent blood counts. The Parkinson Study Group suggested that clozapine, at daily doses of 50 mg or less, is safe and significantly improves drug-induced psychosis without worsening parkinsonism.31 Clozapine may be impractical for elderly patients due to its side effect profile.
Quetiapine is a good alternative to cloza-pine and is less likely to worsen parkinsonian symptoms than other atypical antipsy-chotics.32 Olanzapine and risperidone (Risperdal) are reported to worsen parkinsonian symptoms.33 Not enough data have been published about the efficacy of the newer medications such as ziprasidone (Geodon) and aripiprazole (Abilify) to advocate their routine clinical use.
Rivastigmine (Exelon) was reported to improve hallucinations, sleep disturbance, and caregiver distress in addition to enhancing cognitive performance in advanced Parkinson disease in a small study.34 Burn and colleagues35 reported that rivastigmine was beneficial in patients with dementia associated with Parkinson disease, with or without hallucinations. Efficacy measures were cognitive scales, activities of daily living, behavioral symptoms, and executive and attentional functions. The differences in these measures between rivastigmine and placebo recipients tended to be larger in patients with visual hallucinations than in those without hallucinations. The study was not designed to assess the effect of treatment on psychosis or hallucination.
WHEN PATIENTS WITH MOVEMENT DISORDERS NEED SURGERY
Some of these syndromes can be prevented, especially in patients who are known to have movement disorders and are undergoing surgery.
One problem is stopping oral dopaminergic drugs before the operation. Parkinson disease patients on dopaminergic drugs can develop parkinsonism-hyperpyrexia syndrome or akinetic crisis if the drug is stopped suddenly. Restarting dopaminergic therapy and supportive measures are the main treatments. Patients who have Parkinson disease should receive their usual dose of levodopa, dopamine agonist, or amantadine up until the time of surgery and then again as soon as they awaken in the recovery room.36 That goal can be achieved more easily now that these drugs come in transdermal patches and long-acting formulas.37 Droperidol (Inapsine) and metoclopramide worsen parkinsonism and should be avoided.
Myoclonus is the most common movement disorder seen in the postoperative period. In fact, myoclonic shivering is common as patients awaken from general anesthesia.36 The anesthetic agents etomidate (Amidate) and enflurane (Ethrane) and the opioids fentanyl (Actiq, Duralgesic, Sublimaze) and meperidine (Demerol) can cause myoclonus.38
Occasionally, a patient in the recovery room suddenly develops a neurologic deficit that is inconsistent with the history and physical findings. Psychogenic movement disorders should be considered in the differential diagnosis. Reassurance and occasionally psychiatric intervention are required in these cases.36
IN THE ELDERLY, GO EASY
Polypharmacy is a huge issue in the elderly. Some of the principles in prescribing medications in the elderly can be helpful in preventing movement disorder emergencies:
- Assess the current regimen, including over-the-counter drugs, before prescribing a new drug.
- Begin with a low dose and increase as necessary. “Start low, go slow.”
- Consider the possibility that any new symptoms can be a drug side effect or due to withdrawal of a drug.
- Discuss with the patient or caregiver what kind of side effect to expect and advise him or her to report serious ones.
- Isbister GK, Bowe SJ, Dawson A, Whyte IM. Relative toxicity of selective serotonin reuptake inhibitors (SSRIs) in overdose. J Toxicol Clin Toxicol 2004; 42:277–285.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96:635–642.
- Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000; 79:201–209.
- LoCurto MJ. The serotonin syndrome. Emerg Med Clin North Am 1997; 15( 3):665–675.
- Boyer E, Shannon S. The serotonin syndrome. N Engl J Med 2005; 352:1112–1120.
- Kipps CM, Fung VS, Grattan-Smith P, de Moore GM, Morris JG. Movement disorder emergencies. Mov Disord 2005; 20:322–334.
- Shalev A, Munitz H. The neuroleptic malignant syndrome: agent and host interaction. Acta Psychiatr Scand 1986; 73:337–347.
- Rosebush PI, Stewart TD, Gelenberg AJ. Twenty neuroleptic rechallenges after neuroleptic malignant syndrome in 15 patients. J Clin Psychiatry 1989; 50:295–298.
- Adityanjee , Singh S, Singh G, Ong S. Spectrum concept of neuroleptic malignant syndrome. Br J Psychiatry 1988; 153:107–111.
- Bonuccelli U, Piccini P, Corsini GU, Muratorio A. Apomorphine in malignant syndrome due to levodopa withdrawal. Ital J Neurol Sci 1992; 13:169–170.
- Sato Y, Asoh T, Metoki N, et al. Efficacy of methylprednisolone pulse therapy on neuroleptic malignant syndrome in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2004; 74:574–576.
- Hariz MI, Johansson F. Hardware failure in parkinsonian patients with chronic subthalamic nucleus stimulation is a medical emergency. Mov Disord 2001; 16:166–168.
- Pollera CF, Cognetli F, Nardi M, Mozza D. Sudden death after acute dystonic reaction to high-dose metoclopramide. Lancet 1984; 2:460–461.
- Bateman DN, Rawlins MD, Simpson JM. Extrapyramidal reactions with metoclopramide. Br Med J 1985; 291:930–932.
- Pasricha PJ, Pehlivanov N, Sugumar A, Jankovic J. Drug insight: from disturbed motility to disordered movement—a review of the clinical benefits and medicolegal risks of metoclopramide. Nat Clin Pract Gastroenterol Hepatol 2006; 3:138–148.
- Hu S, Frucht S. Emergency treatment of movement disorders. Curr Treat Options Neurol 2007; 9:103–114.
- Tousi B, Schuele SU, Subramanian T. A 46-year-old woman with rigidity and frequent falls. Cleve Clin J Med 2005; 72:57–63.
- Merlo IM, Occhini A, Pacchetti C, Alfonsi E. Not paralysis, but dystonia causes stridor in multiple system atrophy. Neurology 2002; 58:649–652.
- Silber MH, Levine S. Stridor and death in multiple system atrophy. Mov Disord 2000; 15:699–704.
- Bhatia KP, Marsden CD. The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain 1994; 117:859–876.
- Factor SA, Molho ES. Emergency department presentations of patients with Parkinson’s disease. Am J Emerg Med 2000; 18:209–215.
- Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as a treatment for dyskinesia and motor fluctuations in Parkinson’s disease. Neurology 1998; 50:1323–1326.
- Frucht S, Eidelberg D. Imipenem-induced myoclonus. Mov Disord 1997; 12:621–622.
- Miodownik C, Lerner V, Statsenko N, et al. Vitamin B6 versus mianserin and placebo in acute neuroleptic-induced akathisia: a randomized, double-blind, controlled study. Clin Neuropharmacol 2006; 29:68–72.
- Poyurovsky M, Pashinian A, Weizman R, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry 2006; 59:1071–1077.
- Tousi B, Subramanian T. Hallucinations in Parkinson’s disease: approach and management. Clin Geriatr 2004: 12:19–24.
- Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology 1993; 43:2227–2229.
- Factor SA, Molho ES, Podskalny GD, Brown D. Parkinson’s disease: drug-induced psychiatric states. Adv Neurol 1995; 65:115–138.
- Woodford H, Walker R. Emergency hospital admissions in idiopathic Parkinson’s disease. Mov Disord 2005; 20:1104–1108.
- Tousi B, Frankel M. Olfactory and visual hallucinations in Parkinson’s disease. Parkinsonism Relat Disord 2004; 10:253–254.
- The Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson’s disease. N Engl J Med 1999; 340:757–763.
- Merims D, Balas M, Pertez C, Shabtai H, Giladi N. Rater-blinded, prospective comparison: quetiapine versus clozapine for Parkinson’s disease psychosis. Clin Neuropharmacol 2006; 29:331–337.
- Goetz CG, Blasucci LM, Leurgans S, Pappert EJ. Olanzapine and clozapine: comparative effects on motor function in hallucinating PD patients. Neurology 2000; 55:789–794.
- Reading PJ, Luce AK, McKeith IG. Rivastigmine in the treatment of parkinsonian psychosis and cognitive impairment: preliminary findings from an open trial. Mov Disord 2001; 16:1171–1174.
- Burn D, Emre M, McKeith I, et al. Effects of rivastigmine in patients with and without visual hallucinations in dementia associated with Parkinson’s disease. Mov Disord 2006; 21:1899–1907.
- Frucht SJ. Movement disorder emergencies in the perioperative period. Neurol Clin 2004; 22:379–387.
- Korczyn AD, Reichmann H, Boroojerdi B, et al. Rotigotin trans-dermal system for perioperative administration. J Neural Transm 2007; 114:219–221.
- Gordon MF. Toxin and drug-induced myoclonus. Adv Neurol 2002; 89:49–76.
- Isbister GK, Bowe SJ, Dawson A, Whyte IM. Relative toxicity of selective serotonin reuptake inhibitors (SSRIs) in overdose. J Toxicol Clin Toxicol 2004; 42:277–285.
- Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003; 96:635–642.
- Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000; 79:201–209.
- LoCurto MJ. The serotonin syndrome. Emerg Med Clin North Am 1997; 15( 3):665–675.
- Boyer E, Shannon S. The serotonin syndrome. N Engl J Med 2005; 352:1112–1120.
- Kipps CM, Fung VS, Grattan-Smith P, de Moore GM, Morris JG. Movement disorder emergencies. Mov Disord 2005; 20:322–334.
- Shalev A, Munitz H. The neuroleptic malignant syndrome: agent and host interaction. Acta Psychiatr Scand 1986; 73:337–347.
- Rosebush PI, Stewart TD, Gelenberg AJ. Twenty neuroleptic rechallenges after neuroleptic malignant syndrome in 15 patients. J Clin Psychiatry 1989; 50:295–298.
- Adityanjee , Singh S, Singh G, Ong S. Spectrum concept of neuroleptic malignant syndrome. Br J Psychiatry 1988; 153:107–111.
- Bonuccelli U, Piccini P, Corsini GU, Muratorio A. Apomorphine in malignant syndrome due to levodopa withdrawal. Ital J Neurol Sci 1992; 13:169–170.
- Sato Y, Asoh T, Metoki N, et al. Efficacy of methylprednisolone pulse therapy on neuroleptic malignant syndrome in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2004; 74:574–576.
- Hariz MI, Johansson F. Hardware failure in parkinsonian patients with chronic subthalamic nucleus stimulation is a medical emergency. Mov Disord 2001; 16:166–168.
- Pollera CF, Cognetli F, Nardi M, Mozza D. Sudden death after acute dystonic reaction to high-dose metoclopramide. Lancet 1984; 2:460–461.
- Bateman DN, Rawlins MD, Simpson JM. Extrapyramidal reactions with metoclopramide. Br Med J 1985; 291:930–932.
- Pasricha PJ, Pehlivanov N, Sugumar A, Jankovic J. Drug insight: from disturbed motility to disordered movement—a review of the clinical benefits and medicolegal risks of metoclopramide. Nat Clin Pract Gastroenterol Hepatol 2006; 3:138–148.
- Hu S, Frucht S. Emergency treatment of movement disorders. Curr Treat Options Neurol 2007; 9:103–114.
- Tousi B, Schuele SU, Subramanian T. A 46-year-old woman with rigidity and frequent falls. Cleve Clin J Med 2005; 72:57–63.
- Merlo IM, Occhini A, Pacchetti C, Alfonsi E. Not paralysis, but dystonia causes stridor in multiple system atrophy. Neurology 2002; 58:649–652.
- Silber MH, Levine S. Stridor and death in multiple system atrophy. Mov Disord 2000; 15:699–704.
- Bhatia KP, Marsden CD. The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain 1994; 117:859–876.
- Factor SA, Molho ES. Emergency department presentations of patients with Parkinson’s disease. Am J Emerg Med 2000; 18:209–215.
- Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN. Amantadine as a treatment for dyskinesia and motor fluctuations in Parkinson’s disease. Neurology 1998; 50:1323–1326.
- Frucht S, Eidelberg D. Imipenem-induced myoclonus. Mov Disord 1997; 12:621–622.
- Miodownik C, Lerner V, Statsenko N, et al. Vitamin B6 versus mianserin and placebo in acute neuroleptic-induced akathisia: a randomized, double-blind, controlled study. Clin Neuropharmacol 2006; 29:68–72.
- Poyurovsky M, Pashinian A, Weizman R, et al. Low-dose mirtazapine: a new option in the treatment of antipsychotic-induced akathisia. A randomized, double-blind, placebo- and propranolol-controlled trial. Biol Psychiatry 2006; 59:1071–1077.
- Tousi B, Subramanian T. Hallucinations in Parkinson’s disease: approach and management. Clin Geriatr 2004: 12:19–24.
- Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology 1993; 43:2227–2229.
- Factor SA, Molho ES, Podskalny GD, Brown D. Parkinson’s disease: drug-induced psychiatric states. Adv Neurol 1995; 65:115–138.
- Woodford H, Walker R. Emergency hospital admissions in idiopathic Parkinson’s disease. Mov Disord 2005; 20:1104–1108.
- Tousi B, Frankel M. Olfactory and visual hallucinations in Parkinson’s disease. Parkinsonism Relat Disord 2004; 10:253–254.
- The Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson’s disease. N Engl J Med 1999; 340:757–763.
- Merims D, Balas M, Pertez C, Shabtai H, Giladi N. Rater-blinded, prospective comparison: quetiapine versus clozapine for Parkinson’s disease psychosis. Clin Neuropharmacol 2006; 29:331–337.
- Goetz CG, Blasucci LM, Leurgans S, Pappert EJ. Olanzapine and clozapine: comparative effects on motor function in hallucinating PD patients. Neurology 2000; 55:789–794.
- Reading PJ, Luce AK, McKeith IG. Rivastigmine in the treatment of parkinsonian psychosis and cognitive impairment: preliminary findings from an open trial. Mov Disord 2001; 16:1171–1174.
- Burn D, Emre M, McKeith I, et al. Effects of rivastigmine in patients with and without visual hallucinations in dementia associated with Parkinson’s disease. Mov Disord 2006; 21:1899–1907.
- Frucht SJ. Movement disorder emergencies in the perioperative period. Neurol Clin 2004; 22:379–387.
- Korczyn AD, Reichmann H, Boroojerdi B, et al. Rotigotin trans-dermal system for perioperative administration. J Neural Transm 2007; 114:219–221.
- Gordon MF. Toxin and drug-induced myoclonus. Adv Neurol 2002; 89:49–76.
KEY POINTS
- Supportive measures must be taken immediately to maintain the functions of vital organs.
- Serotonin syndrome, which can cause rigidity or stiffness, can be prevented by avoiding multidrug regimens.
- Withdrawing or decreasing the dose of dopaminergic drugs in patients with Parkinson disease can cause parkinsonism-hyperpyrexia syndrome, a condition similar to neuroleptic malignant syndrome.
- Metoclopramide (Reglan) accounts for nearly one-third of all drug-induced movement disorders. The entire spectrum of drug-induced movement disorders, ranging from subtle to life-threatening, can ensue from its use.
- Complications of Parkinson disease include hallucinations, dementia, depression, psychosis, and sleep disorders.