User login
Clinical Conundrum
A 45‐year‐old man who immigrated to Canada from Ghana at the age of 33 presented with a 2‐year history of progressive cognitive changes. He had bifrontal headache, right‐sided scalp paresthesias, and a 40‐pound weight loss. He was unable to perform his job as an auto parts worker. His wife noticed short‐ and long‐term memory problems and poor concentration. On physical exam he had no focal neurological findings but his score on the Mini‐Mental Status Exam (MMSE) was 23/30, with deficits in attention and recall.
The first important element of this illness is its chronicity. His symptoms progressed slowly over 2 years. Second, aside from his neurological problems, he is an otherwise healthy young, African‐born male. This clinical picture could be the early presentation of a demyelinating, infiltrative, or vascular illness. If vascular, it is more likely a vasculitis than atherosclerotic disease. Malignancy and infection are definitely in the differential, but at this point, I think they are less likely to be the cause, given the tempo of presentation. I would begin my investigations with basic blood work and a computerized tomography (CT) scan of his brain.
A CT scan of the head with contrast demonstrated an enlarged left lateral ventricle with no evidence of obstruction in the foramen of Munro.
The radiological findings of communicating hydrocephalus with normal parenchyma imply a disease that affects the leptomeningeal space. Given that we are looking at an illness that can change cerebral spinal fluid (CSF) flow rather than primary parenchymal disease, demyelinating and vascular illnesses are less likely etiologies, and infiltrative diseases move up on my list. Malignancy and infectious diseases remain in the differential.
He disappeared to follow up for 1 year, during which he returned to Ghana and experienced progressive neurological deterioration, with incontinence, gait instability, and inability to converse clearly and perform activities of daily living. On his return to Canada, an urgent CT scan and magnetic resonance imaging (MRI) of the brain demonstrated ongoing and unchanged hydrocephalus with aqueductal stenosis. A referral was made to a neurosurgeon for insertion of a ventriculoperitoneal shunt. A routine preoperative chest radiograph demonstrated new bilateral upper‐zone reticulonodular changes.
He had no respiratory symptoms, fevers, or lymphadenopathy. His occupational history revealed no exposure to asbestos, silica, farms, or mines. He had no history of either respiratory or neurological illness in the past and no travel other than to Ghana and Toronto. When he immigrated to Toronto, Canada, 12 years before, he had a normal chest radiograph and negative PPD tuberculin skin test.
Many illnesses produce asymptomatic changes on chest x‐ray. Oslerian principles would suggest that we should think of a single diagnosis to explain both nodular lung disease and more than 3 years of a progressive disease affecting the leptomeninges. It is unlikely that tuberculosis, other fungal diseases, or malignancy would result in the chest and brain pathology over a 3‐year period without other sequelae. Sarcoidosis could cause both chronic leptomeningeal changes and the radiographic lung findings. The next steps in investigating this patient should include measurement of angiotensin‐converting enzyme (ACE) and serum calcium and pulmonary function tests. I would ultimately send him for a pathological biopsy of his lung tissue to confirm noncaseating granuloma and exclude infection and malignancy.
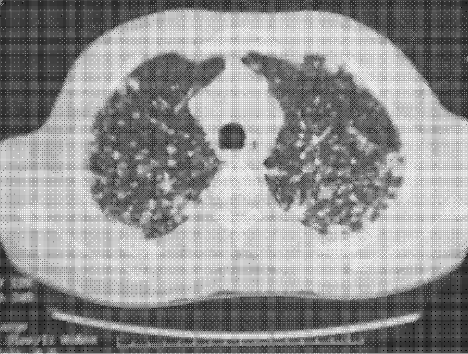
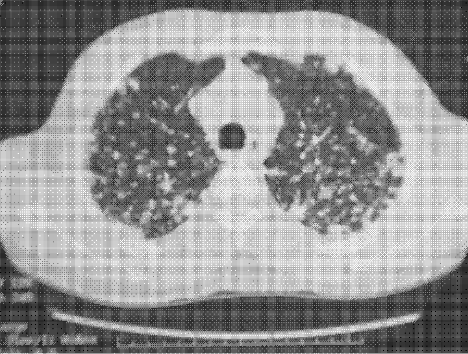
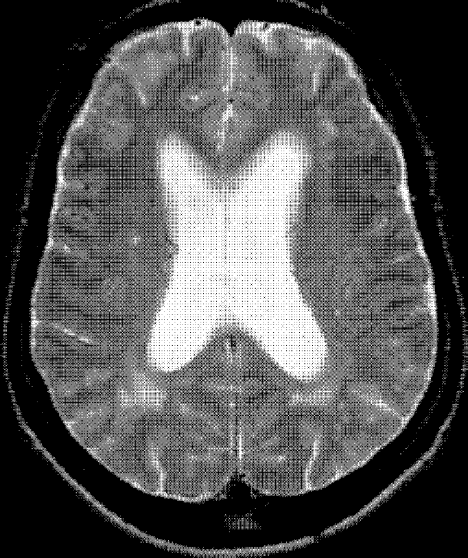
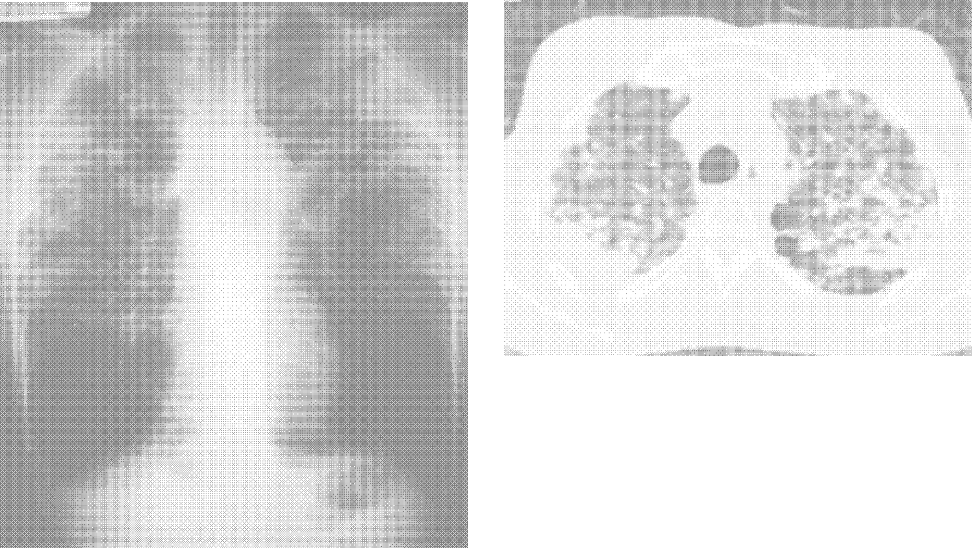
Complete blood count, renal and liver biochemistry, and calcium were normal. An ACE level was elevated at 69 g/L (normal < 40 g/L). A human immunodeficiency virus (HIV‐1 and HIV‐2) test, tuberculin skin test, and syphilis serology were negative. A CT scan of the chest demonstrated bilateral upper‐zone reticulonodular changes and diffuse lymphadenopathy (Fig. 1). Pulmonary function tests (PFTs) demonstrated a forced expiratory volume 1 (FEV1) of 3.4 L (94%), forced vital capacity (FVC) of 4.0 L (83%), an FEV1/FVC of 87%, total lung capacity (TLC) of 92% predicted, and diffusion capacity (DLCO) of 67% predicted. An MRI with gadolinium (Fig. 2) demonstrated hydrocephalus, mild basal leptomeningeal enhancement around the perivascular spaces into the subinsular region, and an increased T2 signal in periventricular white matter.
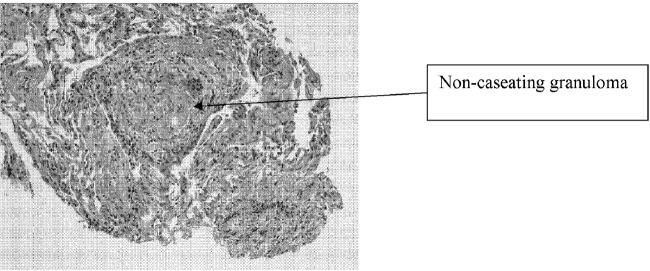
A bronchoscopy with bronchoalveolar lavage and transbronchial biopsies were performed. Pathology (Fig. 3) demonstrated non‐caseating epitheliod granulomas, with negative special stains for acid‐fast bacilli (AFB) and fungus, and negative fungal and AFB cultures of the bronchial alveolar lavage.
With negative tests for infectious causes such as tuberculosis, I think there is now enough evidence that this patient has sarcoidosis involving the lung and leptomeninges. At this point I would start therapy with steroids.
The patient was started on prednisone 40 mg po qd, and his neurological symptoms improved markedly over the course of 1‐2 months, with normalization of his MMSE and a return to cognitive baseline. As his symptoms stabilized with no change in CT imaging, he returned to work, and over the course of 2 years his prednisone dosage was tapered to 10 mg po od. While on prednisone he developed hypertension and hyperglycemia. He continued to have no respiratory symptoms.
He was cognitively at baseline until 20 months later, when he was readmitted to the hospital with a 2‐week history of worsening headache, increased confusion, poor memory, and wandering. His MMSE had deteriorated to 19/30, with deficits again in memory and attention.
First, we can say with reasonable confidence that the diagnosis of sarcoid was correct. His long and sustained response to steroids, plus the absence of the unmasking of an infectious or malignant disease, supports this conclusion. However, he is now exhibiting an apparent relapse that mimics his presentation 3 years earlier. The question is whether he is suffering from a flare of his disease or whether a second illness has occurred. The most obvious second illness is an opportunistic infection after years of steroid use. I would certainly repeat the angiotensin‐converting enzyme and serum calcium tests and repeat the imaging of his lungs and central nervous system. He also warrants a lumbar puncture with CSF culture, stain, and PCR for opportunistic infections. If these studies are inconclusive and do not specifically suggest relapsing sarcoid, I would once again consider biopsy of tissue from either a lung or leptomeninges.
An MRI with gadolinium looked unchanged from the previous one. A lumbar puncture was performed, and his CSF demonstrated 3 WBCs, no RBCs, normal glucose, and elevated protein at 1.17 g/L, and tests for bacteria, TB, fungi, and viruses were all negative. Repeat blood work was unremarkable, and the ACE level was 2 g/L.
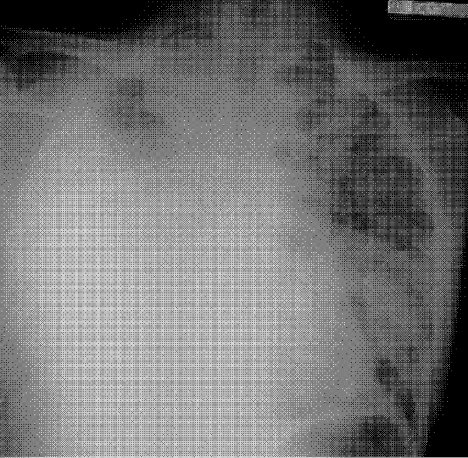
A chest radiograph (Fig. 4a) and CT chest (Fig. 4b) showed marked deterioration, with increased diffuse airspace opacities, interstitial nodularity, and small apical bullae. His PFTs showed some deterioration, with FEV1 2.52 L (73%), FVC 3.29 L (73%), FEV1/FVC 76%, TLC 70% predicted, DLCO 72% predicted. However, he still had no respiratory symptoms.
The changes on lumbar puncture are nonspecific. The ACE level is now very low, making sarcoidosis unlikely but not impossible. The chest imaging shows features, specifically interstitial nodularity, consistent with ongoing or relapsing sarcoidosis, but the extensive apical bullae are not characteristic. My best guess is that this patient's illness is not simply relapsing sarcoid but represents superimposed opportunistic infectious disease. I would not reintroduce steroids without pursuing a definitive diagnosis with tissue pathology.
He was placed on prednisone 60 mg po qd and started on trimethoprim‐sulfamethoxazole for Pneumocystis pneumonia (PCP) prophylaxis. He showed modest improvement in his neurological status. A repeat bronchoscopy was not performed. Four months later he was seen by his pulmonologist. He remained without respiratory symptoms and was neurologically unchanged, and a chest radiograph showed no change. He was continued on prednisone 60 mg po qd.
Three weeks later, he was admitted to the hospital with a 2‐week history of anorexia, fatigue, night sweats, right‐sided pleuritic chest pain with productive cough, increasing dyspnea, and no hemoptysis. On admission he was hypoxic with evidence of respiratory distress, and his chest radiograph showed evidence of new right‐sided airspace disease with an associated large right pleural effusion. Initial labs demonstrated a leukocytosis.
I am now very suspicious that this illness is not relapsed sarcoidosis based on his prior clinical response to high‐dose prednisone and that he currently is showing no neurological improvement. His recent clinical deterioration on this very high dose of prednisone makes me think that opportunistic lung infection or disseminated disease is definitely the cause, although the differential is broad. In addition to the typical viral and bacterial causes of community‐acquired pneumonia, this could be caused by unusual bacterial pathogens, tuberculosis, nontuberculous mycobacteria, or fungal diseases including Candida, Aspergillus and dimorphic fungi. I would begin empiric therapy with antibiotics, obtain pleural fluid for examination and culture, and blood cultures.
The patient was treated with a respiratory fluoroquinolone, and blood and sputum cultures were performed. A right thoracentesis removed 300 cc of yellow exudate, with negative gram stain and initial culture. Over the next 24 hours, the patient deteriorated rapidly, with progressive hypoxia and clinical and radiological (Fig. 5) evidence of acute respiratory distress syndrome (ARDS). He required endotracheal intubation with mechanical ventilation.
He has a progressive illness not responsive to broad‐spectrum antibiotics, and he has deteriorated. At this point it is imperative that he undergo bronchoscopy and transbronchial biopsy.
Bronchoscopy demonstrated secretions from the right lower lobe. Gram stain from a bronchoalveolar lavage from the right lower lobe was negative, and cultures showed no growth after 24 hours. Immediately after bronchoscopy a third‐generation cephalosporin was empirically added. The next day the patient developed hypotension and was started on norepinephrine. Over the subsequent 48 hours, he developed progressive multiorgan failure. Despite multiple vasopressors, high‐frequency oscillator ventilation, broad‐spectrum antimicrobials, and activated protein C, he died in the intensive care unit. At the time of death, all blood cultures were negative, abdominal CT scans showed no intraabdominal infections, and the BAL performed on admission demonstrated negative gram stain, fungal stain, AFB stain, and PCP and no growth from fungal or bacterial cultures.
I think it is an unavoidable conclusion that this man's progressive systemic inflammatory response syndrome and ultimate multiorgan failure was caused by an opportunistic pathogen that was not antibiotic responsive and not identified from the extensive range of infectious disease studies performed. Despite all the negative studies, there still might be either mycobacterial illness or fungal illness. With negative cultures, Candida or Aspergillus infection is unlikely. Other opportunistic fungi like Blastomyces, Histoplasma, and Cryptococcus are certainly in the differential because these organisms can be notoriously difficult to detect on routine surveying such as bronchoalveolar lavage or lumbar puncture. Blastomyces and Histoplasma are both endemic in the area of Canada where the patient resided. I would also keep the zygomycoses in the differential.
Five days after death, fungal culture was reported demonstrating Blastomyces dermatitidis. Postmortem demonstrated disseminated blastomycosis causing severe bilateral pneumonia (Fig. 6a), empyema of right lung, and involvement of the thyroid, heart, liver, spleen, and kidneys. There was also evidence of active CNS blastomycosis involving the meninges and cerebral cortex and diencephalon (Fig. 6b). As well as active blastomycosis, the leptomeninges demonstrated fibrosis and old granulomas that did not contain an organism.

COMMENTARY
This case describes a 45‐year‐old man who presented with chronic cognitive symptoms associated with hydrocephalus. The first step in establishing the diagnosis was made by realizing that a communicating hydrocephalus with no parenchymal CNS disease was highly suggestive of a leptomeningeal process. This narrowed the differential diagnosis to an infiltrative disease affecting the leptomeninges. The next step involved the discovery of an upper‐lobe interstitial lung process, establishing sarcoidosis as the most likely unifying diagnosis. This was confirmed with transbronchial biopsies showing noncaseating granulomas and by the sustained response to treatment with corticosteroids. Unfortunately, after a 2‐year remission, he developed a recurrence of both the neurological and respiratory findings. When his symptoms progressed despite higher doses of corticosteroids, it became apparent that the etiology of his clinical deterioration was not recurrent disease. Instead, the deterioration was caused by disseminated blastomycosis, an opportunistic infection that developed as a result of the immunosuppressants used to treat the sarcoidosis.
With the final diagnosis of blastomycosis, one question about this case becomes: Could it have been blastomycosis and not sarcoid that was responsible for his original neurological presentation? Blastomyces dermatitidis is a thermally dimorphic fungus that causes disease from inhalation of airborne spores found in soil. Areas of North America in which it is endemic include regions bordering the Mississipi and Ohio rivers, as well as the areas bordering the Great Lakes.1 The patient in this case lived in metropolitan Toronto, on Lake Ontario, where blastomycosis is an important yet underreported disease.24 He likely was exposed to blastomyces in Toronto, which in immunocompromised patients may be followed after weeks to months by dissemination to other body sites including the dermis, bones, joints, urogenital system, and, rarely, the central nervous system (CNS) and liver.5 Like sarcoidosis, infection with blastomycosis can produce pathologic evidence of noncaseating granulomatous inflammation. However, as the discussant astutely pointed out, it would be unusual for this patient to have clinically inapparent blastomycosis for almost 2 years while on high‐dose prednisone. The initial diagnosis of sarcoid likely was correct.
CNS disease caused by Blastomyces dermatitidis is quite rare, with only 22 reported cases of meningoencephalitis in the literature.6 As this case demonstrates, CNS blastomycosis is very difficult to diagnose because of the absence of sensitive serologic markers and the difficulty of isolating the organism from blood and cerebrospinal fluid. CSF sampling from lumbar puncture led to its diagnosis in only 2 of the 22 reported cases.7 Furthermore, reliable CSF cultures are usually only obtained via ventricular sampling or tissue biopsy, which itself is limited by the organism's predilection for deep structures of the cerebrum, midbrain, and basal meninges.6 Blastomyces involving the CNS rarely occurs in isolation. In the patient's case, during his neurological deterioration, there was clear radiological evidence of progressive pulmonary pathology despite his being asymptomatic, and as the discussant suggests, pulmonary investigations were warranted.
Pulmonary manifestations of blastomycosis are variable. Acute infections most commonly resemble pneumonia, whereas chronic disease may show reticulonodular changes indistinguishable from sarcoidosis. Severe cases have been shown to progress to respiratory failure with acute respiratory distress syndrome (ARDS).1 The diagnosis is usually established through culture of noninvasive (sensitivity 86%) or bronchoalveolar lavage (sensitivity 92%) specimens.8 However, blastomyces will take between 5 and 30 days to grow in culture.1 In cases where the diagnosis needs to be established quickly, a KOH smear can be done looking for broad‐based budding yeast. Although the yield of this test is lower (0%‐50%), the results can be available within 24 hours.9 As these tests are not always routinely performed, direct communication with the pathologist is recommended if a rapid diagnosis is needed.
The major challenge of this case lay in distinguishing between the recurrence of an old disease and the complications of its treatment. In this case the discussant addresses strategies that might be useful in differentiating recurrent sarcoidosis from an opportunistic infection like blastomycosis. The first issue is the steroid therapy. The exact dose of steroids required to compromise the immune system enough to yield infections is not known. However, in a meta‐analysis of 71 controlled clinical trials performed with steroids, Stuck et. al. were able to show that the occurrence of opportunistic infections depended on both the amount of daily steroid and the cumulative dose.10 Opportunistic infections were unlikely to occur in patients given a mean daily dose of less than 10 mg/day or a cumulative dose of less than 700 mg of prednisone. Although the patient in the present case was only on 10 mg/day of prednisone, his mean daily dose was more than 10 mg/day, and his cumulative dose far exceeded 700 mg. Therefore, an opportunistic infection should have been strongly considered.
The other item used to help distinguish between the 2 diseases was serum angiotensin‐converting enzyme (ACE) level. ACE is an enzyme produced by the epithelial cells of the granulomas in sarcoidosis. ACE alone is inadequate for diagnosis, with a reported sensitivity of 40%‐90%, depending on the population studied and on the definition of normal.1114 Even an ACE level more than twice the normal is not diagnostic for sarcoidosis, with elevated levels found in histoplasmosis, silicosis, tuberculosis, Gaucher's disease, and other disorders.15 Rather than as a diagnostic test, ACE level instead is used to follow disease activity in sarcoidosis, as ACE level often reflects the granuloma burden.1618 The low levels at the initial recurrence suggests the symptoms were not a result of active sarcoid, especially considering that if ACE levels are originally elevated with sarcoidosis, they are almost always elevated again when the disease recurs.14 Normal levels of ACE in sarcoid patients with previously elevated ACE levels should therefore prompt a search for an alternate diagnosis.
This case is an example of the therapy causing a complication that mimics the disease it was intended to cure. When any patient deteriorates while on steroids, the clinician must ask the age‐old question: should more steroids be prescribed or less? As in this case, the answer is not always apparent. Safe decisions in these situations demand awareness of the opportunistic infections endemic to the area and a willingness to perform early invasive procedures (in this case bronchoscopy) to obtain samples to make a definitive diagnosis. By doing so, the devastating chain of events that occurred here hopefully can be avoided.
Acknowledgements
The authors would like to acknowledge Dr. Eleanor Latta and Dr. Serge Jothy, Department of Pathology, St. Michael's Hospital, University of Toronto, for contributing the pathological images.
- ,,.Blastomycosis.Infect Dis Clin North Am.2003;17(1):21,40, vii.
- ,,,,,.Novel cases of blastomycosis acquired in Toronto, Ontario.CMAJ.2000;163:1309–1312.
- ,,,,,.Blastomycosis acquired by three children in Toronto.Can J Infect Dis Med Micro.2002;13(4):259–263.
- ,,, et al.Blastomycosis in Ontario, 1994‐2003.Emerg Infect Dis.2006;12(2):274–279.
- ,,, et al.Epidemiology and clinical spectrum of blastomycosis diagnosed at Manitoba hospitals.Clin Infect Dis.2002;34:1310–1316.
- ,,,.Meningoencephalitis due to Blastomyces dermatitidis: case report and literature review.Mayo Clin Proc.2000;75:403–408.
- ,,,.Chronic blastomycotic meningitis.Am J Med.1981;71:501–505.
- ,.Pulmonary blastomycosis: an appraisal of diagnostic techniques.Chest.2002;121:768–773.
- ,,.Blastomycosis as an etiology of acute lung injury.South Med J.1998;91:861–863.
- ,,.Risk of infectious complications in patients taking glucocorticosteroids.Rev Infect Dis.1989;11:954–963.
- .Elevation of serum angiotensin‐converting‐enzyme (ACE) level in sarcoidosis.Am J Med.1975;59:365–372.
- ,,,.Elevated serum angiotensin I converting enzyme in sarcoidosis.Am Rev Respir Dis.1976;114:525–528.
- ,,.Serum angiotensin‐converting enzyme (SACE) in sarcoidosis and other granulomatous disorders.Lancet.1978;2:1331–1334.
- ,.Serum angiotensin converting enzyme in sarcoidosis: sensitivity and specificity in diagnosis: correlations with disease activity, duration, extra‐thoracic involvement, radiographic type and therapy.Q J Med.1985;55(218):253–270.
- Statement on sarcoidosis.Joint Statement of the American Thoracic Society (ATS), theEuropean Respiratory Society (ERS) and theWorld Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999.Am J Respir Crit Care Med.1999;160:736–755.
- ,,.Value of serial measurement of serum angiotensin converting enzyme in the management of sarcoidosis.Am J Med.1981;70(1):44–50.
- ,,,.Serum angiotensin‐converting enzyme (SACE) activity as an indicator of total body granuloma load and prognosis in sarcoidosis.Sarcoidosis.1987;4(2):142–148.
- ,,.Serum angiotensin converting enzyme in sarcoidosis: clinical significance.Isr J Med Sci.1977;13:1001–1006.
A 45‐year‐old man who immigrated to Canada from Ghana at the age of 33 presented with a 2‐year history of progressive cognitive changes. He had bifrontal headache, right‐sided scalp paresthesias, and a 40‐pound weight loss. He was unable to perform his job as an auto parts worker. His wife noticed short‐ and long‐term memory problems and poor concentration. On physical exam he had no focal neurological findings but his score on the Mini‐Mental Status Exam (MMSE) was 23/30, with deficits in attention and recall.
The first important element of this illness is its chronicity. His symptoms progressed slowly over 2 years. Second, aside from his neurological problems, he is an otherwise healthy young, African‐born male. This clinical picture could be the early presentation of a demyelinating, infiltrative, or vascular illness. If vascular, it is more likely a vasculitis than atherosclerotic disease. Malignancy and infection are definitely in the differential, but at this point, I think they are less likely to be the cause, given the tempo of presentation. I would begin my investigations with basic blood work and a computerized tomography (CT) scan of his brain.
A CT scan of the head with contrast demonstrated an enlarged left lateral ventricle with no evidence of obstruction in the foramen of Munro.
The radiological findings of communicating hydrocephalus with normal parenchyma imply a disease that affects the leptomeningeal space. Given that we are looking at an illness that can change cerebral spinal fluid (CSF) flow rather than primary parenchymal disease, demyelinating and vascular illnesses are less likely etiologies, and infiltrative diseases move up on my list. Malignancy and infectious diseases remain in the differential.
He disappeared to follow up for 1 year, during which he returned to Ghana and experienced progressive neurological deterioration, with incontinence, gait instability, and inability to converse clearly and perform activities of daily living. On his return to Canada, an urgent CT scan and magnetic resonance imaging (MRI) of the brain demonstrated ongoing and unchanged hydrocephalus with aqueductal stenosis. A referral was made to a neurosurgeon for insertion of a ventriculoperitoneal shunt. A routine preoperative chest radiograph demonstrated new bilateral upper‐zone reticulonodular changes.
He had no respiratory symptoms, fevers, or lymphadenopathy. His occupational history revealed no exposure to asbestos, silica, farms, or mines. He had no history of either respiratory or neurological illness in the past and no travel other than to Ghana and Toronto. When he immigrated to Toronto, Canada, 12 years before, he had a normal chest radiograph and negative PPD tuberculin skin test.
Many illnesses produce asymptomatic changes on chest x‐ray. Oslerian principles would suggest that we should think of a single diagnosis to explain both nodular lung disease and more than 3 years of a progressive disease affecting the leptomeninges. It is unlikely that tuberculosis, other fungal diseases, or malignancy would result in the chest and brain pathology over a 3‐year period without other sequelae. Sarcoidosis could cause both chronic leptomeningeal changes and the radiographic lung findings. The next steps in investigating this patient should include measurement of angiotensin‐converting enzyme (ACE) and serum calcium and pulmonary function tests. I would ultimately send him for a pathological biopsy of his lung tissue to confirm noncaseating granuloma and exclude infection and malignancy.
Complete blood count, renal and liver biochemistry, and calcium were normal. An ACE level was elevated at 69 g/L (normal < 40 g/L). A human immunodeficiency virus (HIV‐1 and HIV‐2) test, tuberculin skin test, and syphilis serology were negative. A CT scan of the chest demonstrated bilateral upper‐zone reticulonodular changes and diffuse lymphadenopathy (Fig. 1). Pulmonary function tests (PFTs) demonstrated a forced expiratory volume 1 (FEV1) of 3.4 L (94%), forced vital capacity (FVC) of 4.0 L (83%), an FEV1/FVC of 87%, total lung capacity (TLC) of 92% predicted, and diffusion capacity (DLCO) of 67% predicted. An MRI with gadolinium (Fig. 2) demonstrated hydrocephalus, mild basal leptomeningeal enhancement around the perivascular spaces into the subinsular region, and an increased T2 signal in periventricular white matter.
A bronchoscopy with bronchoalveolar lavage and transbronchial biopsies were performed. Pathology (Fig. 3) demonstrated non‐caseating epitheliod granulomas, with negative special stains for acid‐fast bacilli (AFB) and fungus, and negative fungal and AFB cultures of the bronchial alveolar lavage.
With negative tests for infectious causes such as tuberculosis, I think there is now enough evidence that this patient has sarcoidosis involving the lung and leptomeninges. At this point I would start therapy with steroids.
The patient was started on prednisone 40 mg po qd, and his neurological symptoms improved markedly over the course of 1‐2 months, with normalization of his MMSE and a return to cognitive baseline. As his symptoms stabilized with no change in CT imaging, he returned to work, and over the course of 2 years his prednisone dosage was tapered to 10 mg po od. While on prednisone he developed hypertension and hyperglycemia. He continued to have no respiratory symptoms.
He was cognitively at baseline until 20 months later, when he was readmitted to the hospital with a 2‐week history of worsening headache, increased confusion, poor memory, and wandering. His MMSE had deteriorated to 19/30, with deficits again in memory and attention.
First, we can say with reasonable confidence that the diagnosis of sarcoid was correct. His long and sustained response to steroids, plus the absence of the unmasking of an infectious or malignant disease, supports this conclusion. However, he is now exhibiting an apparent relapse that mimics his presentation 3 years earlier. The question is whether he is suffering from a flare of his disease or whether a second illness has occurred. The most obvious second illness is an opportunistic infection after years of steroid use. I would certainly repeat the angiotensin‐converting enzyme and serum calcium tests and repeat the imaging of his lungs and central nervous system. He also warrants a lumbar puncture with CSF culture, stain, and PCR for opportunistic infections. If these studies are inconclusive and do not specifically suggest relapsing sarcoid, I would once again consider biopsy of tissue from either a lung or leptomeninges.
An MRI with gadolinium looked unchanged from the previous one. A lumbar puncture was performed, and his CSF demonstrated 3 WBCs, no RBCs, normal glucose, and elevated protein at 1.17 g/L, and tests for bacteria, TB, fungi, and viruses were all negative. Repeat blood work was unremarkable, and the ACE level was 2 g/L.
A chest radiograph (Fig. 4a) and CT chest (Fig. 4b) showed marked deterioration, with increased diffuse airspace opacities, interstitial nodularity, and small apical bullae. His PFTs showed some deterioration, with FEV1 2.52 L (73%), FVC 3.29 L (73%), FEV1/FVC 76%, TLC 70% predicted, DLCO 72% predicted. However, he still had no respiratory symptoms.
The changes on lumbar puncture are nonspecific. The ACE level is now very low, making sarcoidosis unlikely but not impossible. The chest imaging shows features, specifically interstitial nodularity, consistent with ongoing or relapsing sarcoidosis, but the extensive apical bullae are not characteristic. My best guess is that this patient's illness is not simply relapsing sarcoid but represents superimposed opportunistic infectious disease. I would not reintroduce steroids without pursuing a definitive diagnosis with tissue pathology.
He was placed on prednisone 60 mg po qd and started on trimethoprim‐sulfamethoxazole for Pneumocystis pneumonia (PCP) prophylaxis. He showed modest improvement in his neurological status. A repeat bronchoscopy was not performed. Four months later he was seen by his pulmonologist. He remained without respiratory symptoms and was neurologically unchanged, and a chest radiograph showed no change. He was continued on prednisone 60 mg po qd.
Three weeks later, he was admitted to the hospital with a 2‐week history of anorexia, fatigue, night sweats, right‐sided pleuritic chest pain with productive cough, increasing dyspnea, and no hemoptysis. On admission he was hypoxic with evidence of respiratory distress, and his chest radiograph showed evidence of new right‐sided airspace disease with an associated large right pleural effusion. Initial labs demonstrated a leukocytosis.
I am now very suspicious that this illness is not relapsed sarcoidosis based on his prior clinical response to high‐dose prednisone and that he currently is showing no neurological improvement. His recent clinical deterioration on this very high dose of prednisone makes me think that opportunistic lung infection or disseminated disease is definitely the cause, although the differential is broad. In addition to the typical viral and bacterial causes of community‐acquired pneumonia, this could be caused by unusual bacterial pathogens, tuberculosis, nontuberculous mycobacteria, or fungal diseases including Candida, Aspergillus and dimorphic fungi. I would begin empiric therapy with antibiotics, obtain pleural fluid for examination and culture, and blood cultures.
The patient was treated with a respiratory fluoroquinolone, and blood and sputum cultures were performed. A right thoracentesis removed 300 cc of yellow exudate, with negative gram stain and initial culture. Over the next 24 hours, the patient deteriorated rapidly, with progressive hypoxia and clinical and radiological (Fig. 5) evidence of acute respiratory distress syndrome (ARDS). He required endotracheal intubation with mechanical ventilation.
He has a progressive illness not responsive to broad‐spectrum antibiotics, and he has deteriorated. At this point it is imperative that he undergo bronchoscopy and transbronchial biopsy.
Bronchoscopy demonstrated secretions from the right lower lobe. Gram stain from a bronchoalveolar lavage from the right lower lobe was negative, and cultures showed no growth after 24 hours. Immediately after bronchoscopy a third‐generation cephalosporin was empirically added. The next day the patient developed hypotension and was started on norepinephrine. Over the subsequent 48 hours, he developed progressive multiorgan failure. Despite multiple vasopressors, high‐frequency oscillator ventilation, broad‐spectrum antimicrobials, and activated protein C, he died in the intensive care unit. At the time of death, all blood cultures were negative, abdominal CT scans showed no intraabdominal infections, and the BAL performed on admission demonstrated negative gram stain, fungal stain, AFB stain, and PCP and no growth from fungal or bacterial cultures.
I think it is an unavoidable conclusion that this man's progressive systemic inflammatory response syndrome and ultimate multiorgan failure was caused by an opportunistic pathogen that was not antibiotic responsive and not identified from the extensive range of infectious disease studies performed. Despite all the negative studies, there still might be either mycobacterial illness or fungal illness. With negative cultures, Candida or Aspergillus infection is unlikely. Other opportunistic fungi like Blastomyces, Histoplasma, and Cryptococcus are certainly in the differential because these organisms can be notoriously difficult to detect on routine surveying such as bronchoalveolar lavage or lumbar puncture. Blastomyces and Histoplasma are both endemic in the area of Canada where the patient resided. I would also keep the zygomycoses in the differential.
Five days after death, fungal culture was reported demonstrating Blastomyces dermatitidis. Postmortem demonstrated disseminated blastomycosis causing severe bilateral pneumonia (Fig. 6a), empyema of right lung, and involvement of the thyroid, heart, liver, spleen, and kidneys. There was also evidence of active CNS blastomycosis involving the meninges and cerebral cortex and diencephalon (Fig. 6b). As well as active blastomycosis, the leptomeninges demonstrated fibrosis and old granulomas that did not contain an organism.
COMMENTARY
This case describes a 45‐year‐old man who presented with chronic cognitive symptoms associated with hydrocephalus. The first step in establishing the diagnosis was made by realizing that a communicating hydrocephalus with no parenchymal CNS disease was highly suggestive of a leptomeningeal process. This narrowed the differential diagnosis to an infiltrative disease affecting the leptomeninges. The next step involved the discovery of an upper‐lobe interstitial lung process, establishing sarcoidosis as the most likely unifying diagnosis. This was confirmed with transbronchial biopsies showing noncaseating granulomas and by the sustained response to treatment with corticosteroids. Unfortunately, after a 2‐year remission, he developed a recurrence of both the neurological and respiratory findings. When his symptoms progressed despite higher doses of corticosteroids, it became apparent that the etiology of his clinical deterioration was not recurrent disease. Instead, the deterioration was caused by disseminated blastomycosis, an opportunistic infection that developed as a result of the immunosuppressants used to treat the sarcoidosis.
With the final diagnosis of blastomycosis, one question about this case becomes: Could it have been blastomycosis and not sarcoid that was responsible for his original neurological presentation? Blastomyces dermatitidis is a thermally dimorphic fungus that causes disease from inhalation of airborne spores found in soil. Areas of North America in which it is endemic include regions bordering the Mississipi and Ohio rivers, as well as the areas bordering the Great Lakes.1 The patient in this case lived in metropolitan Toronto, on Lake Ontario, where blastomycosis is an important yet underreported disease.24 He likely was exposed to blastomyces in Toronto, which in immunocompromised patients may be followed after weeks to months by dissemination to other body sites including the dermis, bones, joints, urogenital system, and, rarely, the central nervous system (CNS) and liver.5 Like sarcoidosis, infection with blastomycosis can produce pathologic evidence of noncaseating granulomatous inflammation. However, as the discussant astutely pointed out, it would be unusual for this patient to have clinically inapparent blastomycosis for almost 2 years while on high‐dose prednisone. The initial diagnosis of sarcoid likely was correct.
CNS disease caused by Blastomyces dermatitidis is quite rare, with only 22 reported cases of meningoencephalitis in the literature.6 As this case demonstrates, CNS blastomycosis is very difficult to diagnose because of the absence of sensitive serologic markers and the difficulty of isolating the organism from blood and cerebrospinal fluid. CSF sampling from lumbar puncture led to its diagnosis in only 2 of the 22 reported cases.7 Furthermore, reliable CSF cultures are usually only obtained via ventricular sampling or tissue biopsy, which itself is limited by the organism's predilection for deep structures of the cerebrum, midbrain, and basal meninges.6 Blastomyces involving the CNS rarely occurs in isolation. In the patient's case, during his neurological deterioration, there was clear radiological evidence of progressive pulmonary pathology despite his being asymptomatic, and as the discussant suggests, pulmonary investigations were warranted.
Pulmonary manifestations of blastomycosis are variable. Acute infections most commonly resemble pneumonia, whereas chronic disease may show reticulonodular changes indistinguishable from sarcoidosis. Severe cases have been shown to progress to respiratory failure with acute respiratory distress syndrome (ARDS).1 The diagnosis is usually established through culture of noninvasive (sensitivity 86%) or bronchoalveolar lavage (sensitivity 92%) specimens.8 However, blastomyces will take between 5 and 30 days to grow in culture.1 In cases where the diagnosis needs to be established quickly, a KOH smear can be done looking for broad‐based budding yeast. Although the yield of this test is lower (0%‐50%), the results can be available within 24 hours.9 As these tests are not always routinely performed, direct communication with the pathologist is recommended if a rapid diagnosis is needed.
The major challenge of this case lay in distinguishing between the recurrence of an old disease and the complications of its treatment. In this case the discussant addresses strategies that might be useful in differentiating recurrent sarcoidosis from an opportunistic infection like blastomycosis. The first issue is the steroid therapy. The exact dose of steroids required to compromise the immune system enough to yield infections is not known. However, in a meta‐analysis of 71 controlled clinical trials performed with steroids, Stuck et. al. were able to show that the occurrence of opportunistic infections depended on both the amount of daily steroid and the cumulative dose.10 Opportunistic infections were unlikely to occur in patients given a mean daily dose of less than 10 mg/day or a cumulative dose of less than 700 mg of prednisone. Although the patient in the present case was only on 10 mg/day of prednisone, his mean daily dose was more than 10 mg/day, and his cumulative dose far exceeded 700 mg. Therefore, an opportunistic infection should have been strongly considered.
The other item used to help distinguish between the 2 diseases was serum angiotensin‐converting enzyme (ACE) level. ACE is an enzyme produced by the epithelial cells of the granulomas in sarcoidosis. ACE alone is inadequate for diagnosis, with a reported sensitivity of 40%‐90%, depending on the population studied and on the definition of normal.1114 Even an ACE level more than twice the normal is not diagnostic for sarcoidosis, with elevated levels found in histoplasmosis, silicosis, tuberculosis, Gaucher's disease, and other disorders.15 Rather than as a diagnostic test, ACE level instead is used to follow disease activity in sarcoidosis, as ACE level often reflects the granuloma burden.1618 The low levels at the initial recurrence suggests the symptoms were not a result of active sarcoid, especially considering that if ACE levels are originally elevated with sarcoidosis, they are almost always elevated again when the disease recurs.14 Normal levels of ACE in sarcoid patients with previously elevated ACE levels should therefore prompt a search for an alternate diagnosis.
This case is an example of the therapy causing a complication that mimics the disease it was intended to cure. When any patient deteriorates while on steroids, the clinician must ask the age‐old question: should more steroids be prescribed or less? As in this case, the answer is not always apparent. Safe decisions in these situations demand awareness of the opportunistic infections endemic to the area and a willingness to perform early invasive procedures (in this case bronchoscopy) to obtain samples to make a definitive diagnosis. By doing so, the devastating chain of events that occurred here hopefully can be avoided.
Acknowledgements
The authors would like to acknowledge Dr. Eleanor Latta and Dr. Serge Jothy, Department of Pathology, St. Michael's Hospital, University of Toronto, for contributing the pathological images.
A 45‐year‐old man who immigrated to Canada from Ghana at the age of 33 presented with a 2‐year history of progressive cognitive changes. He had bifrontal headache, right‐sided scalp paresthesias, and a 40‐pound weight loss. He was unable to perform his job as an auto parts worker. His wife noticed short‐ and long‐term memory problems and poor concentration. On physical exam he had no focal neurological findings but his score on the Mini‐Mental Status Exam (MMSE) was 23/30, with deficits in attention and recall.
The first important element of this illness is its chronicity. His symptoms progressed slowly over 2 years. Second, aside from his neurological problems, he is an otherwise healthy young, African‐born male. This clinical picture could be the early presentation of a demyelinating, infiltrative, or vascular illness. If vascular, it is more likely a vasculitis than atherosclerotic disease. Malignancy and infection are definitely in the differential, but at this point, I think they are less likely to be the cause, given the tempo of presentation. I would begin my investigations with basic blood work and a computerized tomography (CT) scan of his brain.
A CT scan of the head with contrast demonstrated an enlarged left lateral ventricle with no evidence of obstruction in the foramen of Munro.
The radiological findings of communicating hydrocephalus with normal parenchyma imply a disease that affects the leptomeningeal space. Given that we are looking at an illness that can change cerebral spinal fluid (CSF) flow rather than primary parenchymal disease, demyelinating and vascular illnesses are less likely etiologies, and infiltrative diseases move up on my list. Malignancy and infectious diseases remain in the differential.
He disappeared to follow up for 1 year, during which he returned to Ghana and experienced progressive neurological deterioration, with incontinence, gait instability, and inability to converse clearly and perform activities of daily living. On his return to Canada, an urgent CT scan and magnetic resonance imaging (MRI) of the brain demonstrated ongoing and unchanged hydrocephalus with aqueductal stenosis. A referral was made to a neurosurgeon for insertion of a ventriculoperitoneal shunt. A routine preoperative chest radiograph demonstrated new bilateral upper‐zone reticulonodular changes.
He had no respiratory symptoms, fevers, or lymphadenopathy. His occupational history revealed no exposure to asbestos, silica, farms, or mines. He had no history of either respiratory or neurological illness in the past and no travel other than to Ghana and Toronto. When he immigrated to Toronto, Canada, 12 years before, he had a normal chest radiograph and negative PPD tuberculin skin test.
Many illnesses produce asymptomatic changes on chest x‐ray. Oslerian principles would suggest that we should think of a single diagnosis to explain both nodular lung disease and more than 3 years of a progressive disease affecting the leptomeninges. It is unlikely that tuberculosis, other fungal diseases, or malignancy would result in the chest and brain pathology over a 3‐year period without other sequelae. Sarcoidosis could cause both chronic leptomeningeal changes and the radiographic lung findings. The next steps in investigating this patient should include measurement of angiotensin‐converting enzyme (ACE) and serum calcium and pulmonary function tests. I would ultimately send him for a pathological biopsy of his lung tissue to confirm noncaseating granuloma and exclude infection and malignancy.
Complete blood count, renal and liver biochemistry, and calcium were normal. An ACE level was elevated at 69 g/L (normal < 40 g/L). A human immunodeficiency virus (HIV‐1 and HIV‐2) test, tuberculin skin test, and syphilis serology were negative. A CT scan of the chest demonstrated bilateral upper‐zone reticulonodular changes and diffuse lymphadenopathy (Fig. 1). Pulmonary function tests (PFTs) demonstrated a forced expiratory volume 1 (FEV1) of 3.4 L (94%), forced vital capacity (FVC) of 4.0 L (83%), an FEV1/FVC of 87%, total lung capacity (TLC) of 92% predicted, and diffusion capacity (DLCO) of 67% predicted. An MRI with gadolinium (Fig. 2) demonstrated hydrocephalus, mild basal leptomeningeal enhancement around the perivascular spaces into the subinsular region, and an increased T2 signal in periventricular white matter.
A bronchoscopy with bronchoalveolar lavage and transbronchial biopsies were performed. Pathology (Fig. 3) demonstrated non‐caseating epitheliod granulomas, with negative special stains for acid‐fast bacilli (AFB) and fungus, and negative fungal and AFB cultures of the bronchial alveolar lavage.
With negative tests for infectious causes such as tuberculosis, I think there is now enough evidence that this patient has sarcoidosis involving the lung and leptomeninges. At this point I would start therapy with steroids.
The patient was started on prednisone 40 mg po qd, and his neurological symptoms improved markedly over the course of 1‐2 months, with normalization of his MMSE and a return to cognitive baseline. As his symptoms stabilized with no change in CT imaging, he returned to work, and over the course of 2 years his prednisone dosage was tapered to 10 mg po od. While on prednisone he developed hypertension and hyperglycemia. He continued to have no respiratory symptoms.
He was cognitively at baseline until 20 months later, when he was readmitted to the hospital with a 2‐week history of worsening headache, increased confusion, poor memory, and wandering. His MMSE had deteriorated to 19/30, with deficits again in memory and attention.
First, we can say with reasonable confidence that the diagnosis of sarcoid was correct. His long and sustained response to steroids, plus the absence of the unmasking of an infectious or malignant disease, supports this conclusion. However, he is now exhibiting an apparent relapse that mimics his presentation 3 years earlier. The question is whether he is suffering from a flare of his disease or whether a second illness has occurred. The most obvious second illness is an opportunistic infection after years of steroid use. I would certainly repeat the angiotensin‐converting enzyme and serum calcium tests and repeat the imaging of his lungs and central nervous system. He also warrants a lumbar puncture with CSF culture, stain, and PCR for opportunistic infections. If these studies are inconclusive and do not specifically suggest relapsing sarcoid, I would once again consider biopsy of tissue from either a lung or leptomeninges.
An MRI with gadolinium looked unchanged from the previous one. A lumbar puncture was performed, and his CSF demonstrated 3 WBCs, no RBCs, normal glucose, and elevated protein at 1.17 g/L, and tests for bacteria, TB, fungi, and viruses were all negative. Repeat blood work was unremarkable, and the ACE level was 2 g/L.
A chest radiograph (Fig. 4a) and CT chest (Fig. 4b) showed marked deterioration, with increased diffuse airspace opacities, interstitial nodularity, and small apical bullae. His PFTs showed some deterioration, with FEV1 2.52 L (73%), FVC 3.29 L (73%), FEV1/FVC 76%, TLC 70% predicted, DLCO 72% predicted. However, he still had no respiratory symptoms.
The changes on lumbar puncture are nonspecific. The ACE level is now very low, making sarcoidosis unlikely but not impossible. The chest imaging shows features, specifically interstitial nodularity, consistent with ongoing or relapsing sarcoidosis, but the extensive apical bullae are not characteristic. My best guess is that this patient's illness is not simply relapsing sarcoid but represents superimposed opportunistic infectious disease. I would not reintroduce steroids without pursuing a definitive diagnosis with tissue pathology.
He was placed on prednisone 60 mg po qd and started on trimethoprim‐sulfamethoxazole for Pneumocystis pneumonia (PCP) prophylaxis. He showed modest improvement in his neurological status. A repeat bronchoscopy was not performed. Four months later he was seen by his pulmonologist. He remained without respiratory symptoms and was neurologically unchanged, and a chest radiograph showed no change. He was continued on prednisone 60 mg po qd.
Three weeks later, he was admitted to the hospital with a 2‐week history of anorexia, fatigue, night sweats, right‐sided pleuritic chest pain with productive cough, increasing dyspnea, and no hemoptysis. On admission he was hypoxic with evidence of respiratory distress, and his chest radiograph showed evidence of new right‐sided airspace disease with an associated large right pleural effusion. Initial labs demonstrated a leukocytosis.
I am now very suspicious that this illness is not relapsed sarcoidosis based on his prior clinical response to high‐dose prednisone and that he currently is showing no neurological improvement. His recent clinical deterioration on this very high dose of prednisone makes me think that opportunistic lung infection or disseminated disease is definitely the cause, although the differential is broad. In addition to the typical viral and bacterial causes of community‐acquired pneumonia, this could be caused by unusual bacterial pathogens, tuberculosis, nontuberculous mycobacteria, or fungal diseases including Candida, Aspergillus and dimorphic fungi. I would begin empiric therapy with antibiotics, obtain pleural fluid for examination and culture, and blood cultures.
The patient was treated with a respiratory fluoroquinolone, and blood and sputum cultures were performed. A right thoracentesis removed 300 cc of yellow exudate, with negative gram stain and initial culture. Over the next 24 hours, the patient deteriorated rapidly, with progressive hypoxia and clinical and radiological (Fig. 5) evidence of acute respiratory distress syndrome (ARDS). He required endotracheal intubation with mechanical ventilation.
He has a progressive illness not responsive to broad‐spectrum antibiotics, and he has deteriorated. At this point it is imperative that he undergo bronchoscopy and transbronchial biopsy.
Bronchoscopy demonstrated secretions from the right lower lobe. Gram stain from a bronchoalveolar lavage from the right lower lobe was negative, and cultures showed no growth after 24 hours. Immediately after bronchoscopy a third‐generation cephalosporin was empirically added. The next day the patient developed hypotension and was started on norepinephrine. Over the subsequent 48 hours, he developed progressive multiorgan failure. Despite multiple vasopressors, high‐frequency oscillator ventilation, broad‐spectrum antimicrobials, and activated protein C, he died in the intensive care unit. At the time of death, all blood cultures were negative, abdominal CT scans showed no intraabdominal infections, and the BAL performed on admission demonstrated negative gram stain, fungal stain, AFB stain, and PCP and no growth from fungal or bacterial cultures.
I think it is an unavoidable conclusion that this man's progressive systemic inflammatory response syndrome and ultimate multiorgan failure was caused by an opportunistic pathogen that was not antibiotic responsive and not identified from the extensive range of infectious disease studies performed. Despite all the negative studies, there still might be either mycobacterial illness or fungal illness. With negative cultures, Candida or Aspergillus infection is unlikely. Other opportunistic fungi like Blastomyces, Histoplasma, and Cryptococcus are certainly in the differential because these organisms can be notoriously difficult to detect on routine surveying such as bronchoalveolar lavage or lumbar puncture. Blastomyces and Histoplasma are both endemic in the area of Canada where the patient resided. I would also keep the zygomycoses in the differential.
Five days after death, fungal culture was reported demonstrating Blastomyces dermatitidis. Postmortem demonstrated disseminated blastomycosis causing severe bilateral pneumonia (Fig. 6a), empyema of right lung, and involvement of the thyroid, heart, liver, spleen, and kidneys. There was also evidence of active CNS blastomycosis involving the meninges and cerebral cortex and diencephalon (Fig. 6b). As well as active blastomycosis, the leptomeninges demonstrated fibrosis and old granulomas that did not contain an organism.
COMMENTARY
This case describes a 45‐year‐old man who presented with chronic cognitive symptoms associated with hydrocephalus. The first step in establishing the diagnosis was made by realizing that a communicating hydrocephalus with no parenchymal CNS disease was highly suggestive of a leptomeningeal process. This narrowed the differential diagnosis to an infiltrative disease affecting the leptomeninges. The next step involved the discovery of an upper‐lobe interstitial lung process, establishing sarcoidosis as the most likely unifying diagnosis. This was confirmed with transbronchial biopsies showing noncaseating granulomas and by the sustained response to treatment with corticosteroids. Unfortunately, after a 2‐year remission, he developed a recurrence of both the neurological and respiratory findings. When his symptoms progressed despite higher doses of corticosteroids, it became apparent that the etiology of his clinical deterioration was not recurrent disease. Instead, the deterioration was caused by disseminated blastomycosis, an opportunistic infection that developed as a result of the immunosuppressants used to treat the sarcoidosis.
With the final diagnosis of blastomycosis, one question about this case becomes: Could it have been blastomycosis and not sarcoid that was responsible for his original neurological presentation? Blastomyces dermatitidis is a thermally dimorphic fungus that causes disease from inhalation of airborne spores found in soil. Areas of North America in which it is endemic include regions bordering the Mississipi and Ohio rivers, as well as the areas bordering the Great Lakes.1 The patient in this case lived in metropolitan Toronto, on Lake Ontario, where blastomycosis is an important yet underreported disease.24 He likely was exposed to blastomyces in Toronto, which in immunocompromised patients may be followed after weeks to months by dissemination to other body sites including the dermis, bones, joints, urogenital system, and, rarely, the central nervous system (CNS) and liver.5 Like sarcoidosis, infection with blastomycosis can produce pathologic evidence of noncaseating granulomatous inflammation. However, as the discussant astutely pointed out, it would be unusual for this patient to have clinically inapparent blastomycosis for almost 2 years while on high‐dose prednisone. The initial diagnosis of sarcoid likely was correct.
CNS disease caused by Blastomyces dermatitidis is quite rare, with only 22 reported cases of meningoencephalitis in the literature.6 As this case demonstrates, CNS blastomycosis is very difficult to diagnose because of the absence of sensitive serologic markers and the difficulty of isolating the organism from blood and cerebrospinal fluid. CSF sampling from lumbar puncture led to its diagnosis in only 2 of the 22 reported cases.7 Furthermore, reliable CSF cultures are usually only obtained via ventricular sampling or tissue biopsy, which itself is limited by the organism's predilection for deep structures of the cerebrum, midbrain, and basal meninges.6 Blastomyces involving the CNS rarely occurs in isolation. In the patient's case, during his neurological deterioration, there was clear radiological evidence of progressive pulmonary pathology despite his being asymptomatic, and as the discussant suggests, pulmonary investigations were warranted.
Pulmonary manifestations of blastomycosis are variable. Acute infections most commonly resemble pneumonia, whereas chronic disease may show reticulonodular changes indistinguishable from sarcoidosis. Severe cases have been shown to progress to respiratory failure with acute respiratory distress syndrome (ARDS).1 The diagnosis is usually established through culture of noninvasive (sensitivity 86%) or bronchoalveolar lavage (sensitivity 92%) specimens.8 However, blastomyces will take between 5 and 30 days to grow in culture.1 In cases where the diagnosis needs to be established quickly, a KOH smear can be done looking for broad‐based budding yeast. Although the yield of this test is lower (0%‐50%), the results can be available within 24 hours.9 As these tests are not always routinely performed, direct communication with the pathologist is recommended if a rapid diagnosis is needed.
The major challenge of this case lay in distinguishing between the recurrence of an old disease and the complications of its treatment. In this case the discussant addresses strategies that might be useful in differentiating recurrent sarcoidosis from an opportunistic infection like blastomycosis. The first issue is the steroid therapy. The exact dose of steroids required to compromise the immune system enough to yield infections is not known. However, in a meta‐analysis of 71 controlled clinical trials performed with steroids, Stuck et. al. were able to show that the occurrence of opportunistic infections depended on both the amount of daily steroid and the cumulative dose.10 Opportunistic infections were unlikely to occur in patients given a mean daily dose of less than 10 mg/day or a cumulative dose of less than 700 mg of prednisone. Although the patient in the present case was only on 10 mg/day of prednisone, his mean daily dose was more than 10 mg/day, and his cumulative dose far exceeded 700 mg. Therefore, an opportunistic infection should have been strongly considered.
The other item used to help distinguish between the 2 diseases was serum angiotensin‐converting enzyme (ACE) level. ACE is an enzyme produced by the epithelial cells of the granulomas in sarcoidosis. ACE alone is inadequate for diagnosis, with a reported sensitivity of 40%‐90%, depending on the population studied and on the definition of normal.1114 Even an ACE level more than twice the normal is not diagnostic for sarcoidosis, with elevated levels found in histoplasmosis, silicosis, tuberculosis, Gaucher's disease, and other disorders.15 Rather than as a diagnostic test, ACE level instead is used to follow disease activity in sarcoidosis, as ACE level often reflects the granuloma burden.1618 The low levels at the initial recurrence suggests the symptoms were not a result of active sarcoid, especially considering that if ACE levels are originally elevated with sarcoidosis, they are almost always elevated again when the disease recurs.14 Normal levels of ACE in sarcoid patients with previously elevated ACE levels should therefore prompt a search for an alternate diagnosis.
This case is an example of the therapy causing a complication that mimics the disease it was intended to cure. When any patient deteriorates while on steroids, the clinician must ask the age‐old question: should more steroids be prescribed or less? As in this case, the answer is not always apparent. Safe decisions in these situations demand awareness of the opportunistic infections endemic to the area and a willingness to perform early invasive procedures (in this case bronchoscopy) to obtain samples to make a definitive diagnosis. By doing so, the devastating chain of events that occurred here hopefully can be avoided.
Acknowledgements
The authors would like to acknowledge Dr. Eleanor Latta and Dr. Serge Jothy, Department of Pathology, St. Michael's Hospital, University of Toronto, for contributing the pathological images.
- ,,.Blastomycosis.Infect Dis Clin North Am.2003;17(1):21,40, vii.
- ,,,,,.Novel cases of blastomycosis acquired in Toronto, Ontario.CMAJ.2000;163:1309–1312.
- ,,,,,.Blastomycosis acquired by three children in Toronto.Can J Infect Dis Med Micro.2002;13(4):259–263.
- ,,, et al.Blastomycosis in Ontario, 1994‐2003.Emerg Infect Dis.2006;12(2):274–279.
- ,,, et al.Epidemiology and clinical spectrum of blastomycosis diagnosed at Manitoba hospitals.Clin Infect Dis.2002;34:1310–1316.
- ,,,.Meningoencephalitis due to Blastomyces dermatitidis: case report and literature review.Mayo Clin Proc.2000;75:403–408.
- ,,,.Chronic blastomycotic meningitis.Am J Med.1981;71:501–505.
- ,.Pulmonary blastomycosis: an appraisal of diagnostic techniques.Chest.2002;121:768–773.
- ,,.Blastomycosis as an etiology of acute lung injury.South Med J.1998;91:861–863.
- ,,.Risk of infectious complications in patients taking glucocorticosteroids.Rev Infect Dis.1989;11:954–963.
- .Elevation of serum angiotensin‐converting‐enzyme (ACE) level in sarcoidosis.Am J Med.1975;59:365–372.
- ,,,.Elevated serum angiotensin I converting enzyme in sarcoidosis.Am Rev Respir Dis.1976;114:525–528.
- ,,.Serum angiotensin‐converting enzyme (SACE) in sarcoidosis and other granulomatous disorders.Lancet.1978;2:1331–1334.
- ,.Serum angiotensin converting enzyme in sarcoidosis: sensitivity and specificity in diagnosis: correlations with disease activity, duration, extra‐thoracic involvement, radiographic type and therapy.Q J Med.1985;55(218):253–270.
- Statement on sarcoidosis.Joint Statement of the American Thoracic Society (ATS), theEuropean Respiratory Society (ERS) and theWorld Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999.Am J Respir Crit Care Med.1999;160:736–755.
- ,,.Value of serial measurement of serum angiotensin converting enzyme in the management of sarcoidosis.Am J Med.1981;70(1):44–50.
- ,,,.Serum angiotensin‐converting enzyme (SACE) activity as an indicator of total body granuloma load and prognosis in sarcoidosis.Sarcoidosis.1987;4(2):142–148.
- ,,.Serum angiotensin converting enzyme in sarcoidosis: clinical significance.Isr J Med Sci.1977;13:1001–1006.
- ,,.Blastomycosis.Infect Dis Clin North Am.2003;17(1):21,40, vii.
- ,,,,,.Novel cases of blastomycosis acquired in Toronto, Ontario.CMAJ.2000;163:1309–1312.
- ,,,,,.Blastomycosis acquired by three children in Toronto.Can J Infect Dis Med Micro.2002;13(4):259–263.
- ,,, et al.Blastomycosis in Ontario, 1994‐2003.Emerg Infect Dis.2006;12(2):274–279.
- ,,, et al.Epidemiology and clinical spectrum of blastomycosis diagnosed at Manitoba hospitals.Clin Infect Dis.2002;34:1310–1316.
- ,,,.Meningoencephalitis due to Blastomyces dermatitidis: case report and literature review.Mayo Clin Proc.2000;75:403–408.
- ,,,.Chronic blastomycotic meningitis.Am J Med.1981;71:501–505.
- ,.Pulmonary blastomycosis: an appraisal of diagnostic techniques.Chest.2002;121:768–773.
- ,,.Blastomycosis as an etiology of acute lung injury.South Med J.1998;91:861–863.
- ,,.Risk of infectious complications in patients taking glucocorticosteroids.Rev Infect Dis.1989;11:954–963.
- .Elevation of serum angiotensin‐converting‐enzyme (ACE) level in sarcoidosis.Am J Med.1975;59:365–372.
- ,,,.Elevated serum angiotensin I converting enzyme in sarcoidosis.Am Rev Respir Dis.1976;114:525–528.
- ,,.Serum angiotensin‐converting enzyme (SACE) in sarcoidosis and other granulomatous disorders.Lancet.1978;2:1331–1334.
- ,.Serum angiotensin converting enzyme in sarcoidosis: sensitivity and specificity in diagnosis: correlations with disease activity, duration, extra‐thoracic involvement, radiographic type and therapy.Q J Med.1985;55(218):253–270.
- Statement on sarcoidosis.Joint Statement of the American Thoracic Society (ATS), theEuropean Respiratory Society (ERS) and theWorld Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999.Am J Respir Crit Care Med.1999;160:736–755.
- ,,.Value of serial measurement of serum angiotensin converting enzyme in the management of sarcoidosis.Am J Med.1981;70(1):44–50.
- ,,,.Serum angiotensin‐converting enzyme (SACE) activity as an indicator of total body granuloma load and prognosis in sarcoidosis.Sarcoidosis.1987;4(2):142–148.
- ,,.Serum angiotensin converting enzyme in sarcoidosis: clinical significance.Isr J Med Sci.1977;13:1001–1006.