User login
Autoantibody-mediated encephalitis: Not just paraneoplastic, not just limbic, and not untreatable
A 79-year-old woman with a history of breast cancer in remission and hypertension presented to a local emergency department because of subacute memory loss and compulsive shopping. Her serum sodium concentration was 127 mmol/L (reference range 132–148). Computed tomography (CT) and magnetic resonance imaging (MRI) of the brain were normal, and she was sent home.
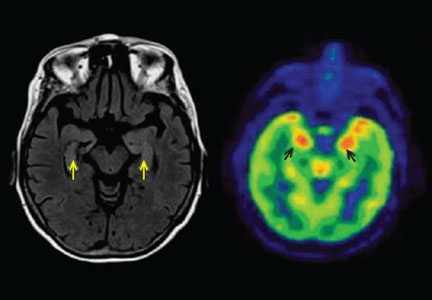
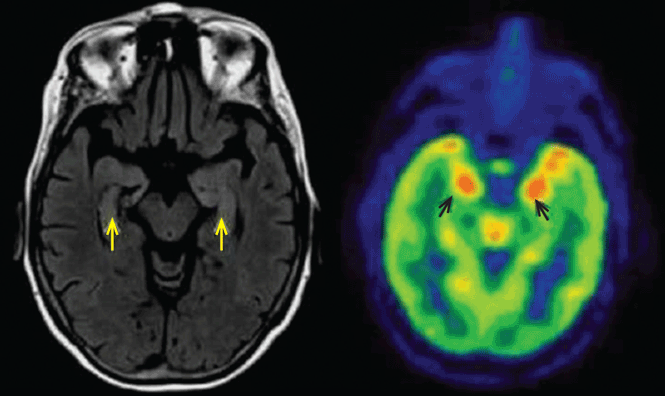
Three days later, she experienced a generalized tonic-clonic seizure that evolved into status epilepticus. She was intubated and admitted to the intensive care unit. Cerebrospinal fluid analysis was normal, and infectious causes of encephalitis were ruled out. MRI showed increased signal in both hippocampi (Figure 1). Her seizures were refractory to treatment, and she was given pentobarbital to induce a coma.
Serum evaluation of neuronal antibodies revealed elevated titers of the voltage-gated potassium channel (VGKC) complex antibody, with subsequent subtyping confirming the leucine-rich glioma-inactivated protein 1 (LGI1) protein as the antigenic target.

She received a 5-day course of intravenous immunoglobulin and methylprednisolone, pentobarbital was withdrawn, and the seizures did not recur, but weeks later she remained comatose. Positron emission tomography (PET) of the brain revealed hypermetabolism in the medial and anterior aspects of both temporal lobes. She underwent five sessions of plasma exchange, after which she began to improve and follow commands. She was ultimately discharged to an acute rehabilitation facility after a 4-week hospital stay.
She received infusions of intravenous immunoglobulin twice a month for 6 months. At her last follow-up visit, she was seizure-free and neurologically intact except for mild inattention.
NEWLY RECOGNIZED DISEASES
Although autoantibody-mediated encephalitic syndromes were first described more than 50 years ago,1,2 their autoimmune basis was not recognized until the early 1980s.3 In the past 10 years, a flood of novel clinical syndromes associated with neuronal autoantibodies has been described that may be markedly improved or even completely resolved with immunotherapy. In cases of unexplained seizure, encephalitis, or acute-onset psychiatric syndromes, suspecting these syndromes can lead to diagnosis, treatment, and a good outcome.
This review describes the key clinical autoantibody-mediated encephalitic syndromes, explains the better-characterized antibody associations, and discusses their diagnosis and treatment.
CLASSIFIED ANATOMICALLY, IMMUNOLOGICALLY, OR EPONYMOUSLY
Autoantibody-mediated encephalitis is also known as autoimmune-mediated encephalitis, autoimmune-mediated limbic encephalitis, and autoimmune synaptic encephalitis.
How to categorize these syndromes is still in flux: they can be listed by the area of the brain affected, the antibody involved, or the name of the discoverer (eg, Morvan syndrome).
Autoantibodies identified in autoimmune encephalitis fall under two broad categories:
- Those targeting intracellular (intranuclear or intracytoplasmic) antigens; the syndromes they cause are more likely to be paraneoplastic and less responsive to immunotherapy
- Those targeting antigens on the neuronal surface: the syndromes they cause are less likely to be paraneoplastic and are more responsive to immunotherapy.4
SYNDROMES DEFINED BY BRAIN AREA AFFECTED
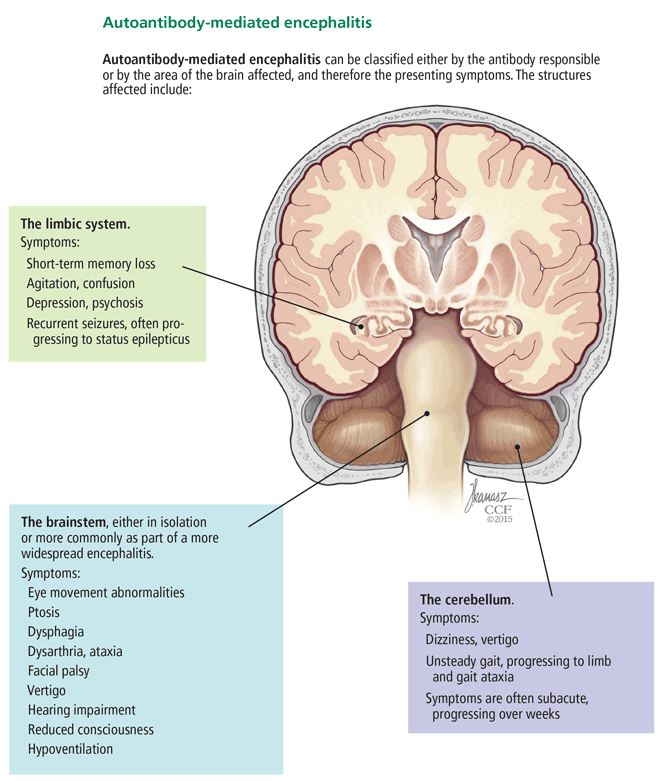
Below, we provide examples of neurologic syndromes of autoantibody-mediated encephalitis according to the region of the brain most affected, ie, the limbic system, the brainstem, or the cerebellum (Figure 2).
LIMBIC ENCEPHALITIS
Memory loss, behavioral changes, seizures
Patients with limbic encephalitis (such as the patient described in the vignette above) present with symptoms attributed to dysfunction of mesial temporal lobe structures, most notably the hippocampus. Prominent symptoms include short-term memory loss, behavioral disturbances such as agitation and confusion, and psychiatric problems such as depression and psychosis. Recurrent seizures are a salient feature and, not uncommonly, progress to status epilepticus.
Antibodies are not all cancer-associated
Cerebrospinal fluid analysis can be normal or show abnormalities suggesting immune activation, eg, slight pleocytosis, elevated protein, increased immunoglobulin G synthesis, and oligoclonal banding.5
In many cases, an autoantibody is found in the blood or in the cerebrospinal fluid. Some patients may express more than one autoantibody, so the traditional view of “one antibody, one syndrome” is incorrect.
Although initially identified as a rare paraneoplastic disorder, limbic encephalitis sometimes occurs in the absence of malignancy.
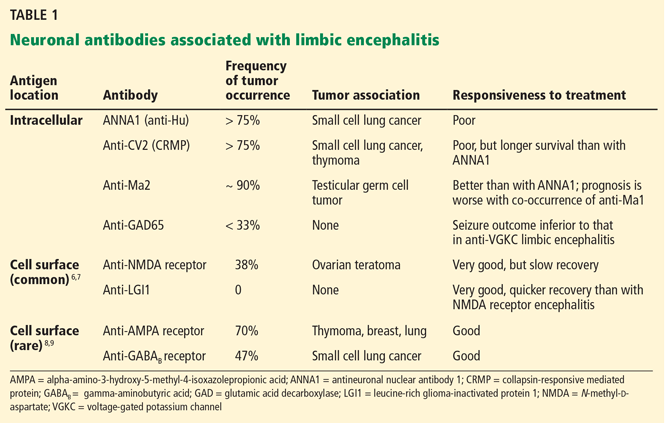
Multiple antibodies have been linked to the syndrome (Table 1).6–9 The “classic” antibodies initially found in paraneoplastic forms are now generally viewed as nonpathogenic, in part because they are directed against intracellular antigens. Neuronal injury in paraneoplastic limbic encephalitis is believed to be mediated by cytotoxic T lymphocytes, with neuronal autoantibodies being produced after the injury.4 Recently defined antibodies, such as those targeting the N-methyl-d-aspartate (NMDA) receptor6 and the LGI1 protein,7 are now understood to be common causes of limbic encephalitis.
Imaging usually shows limbic focal changes
Structural MRI or functional fluorodeoxyglucose (FDG)-PET imaging may show focal changes in limbic system structures, such as the mesial temporal lobes. It is now recognized that other cortical areas may be involved, and the term “limbic encephalitis” may give way to “cortical” or “focal encephalitis.”
In about 60% of patients, MRI shows hyperintense fluid-attenuated inversion recovery (FLAIR) or T2 signal changes in the mesial temporal lobes, likely reflecting inflammatory changes.4,10,11 On FDG-PET, hypermetabolism may be observed in the mesial temporal lobes early in the disease despite normal findings on MRI.12 Hypometabolism, either diffuse or localized to the mesial temporal lobes, eventually sets in, likely reflecting cytotoxic injury in the aftermath of prolonged inflammation or seizures.
Consider other causes
Before diagnosing limbic encephalitis, it is essential to evaluate for infectious meningoencephalitis, especially herpes simplex viral encephalitis. Thiamine deficiency (Wernicke encephalopathy), drug intoxication, prion disease, Hashimoto encephalopathy, tumor, and subclinical status epilepticus should also be considered. Some of these conditions are associated with the same neuronal autoantibodies detected in limbic encephalitis. Further complicating the picture, case reports have shown the presence of serum neuronal autoantibodies—VGKC complex13–15 and NMDA-receptor antibodies16,17—in confirmed cases of prion disease. In addition, adequately treated herpes simplex viral encephalitis can precipitate the production of NMDA-receptor antibodies and their characteristic syndrome.18–20
BRAINSTEM ENCEPHALITIS
The brainstem—the midbrain, pons, and medulla—can be affected, either in isolation or more commonly as part of a more widespread autoantibody-mediated encephalitis. Symptoms and signs include eye movement abnormalities, ptosis, dysphagia, dysarthria, ataxia, facial palsy, vertigo, hearing impairment, reduced consciousness, and hypoventilation.21
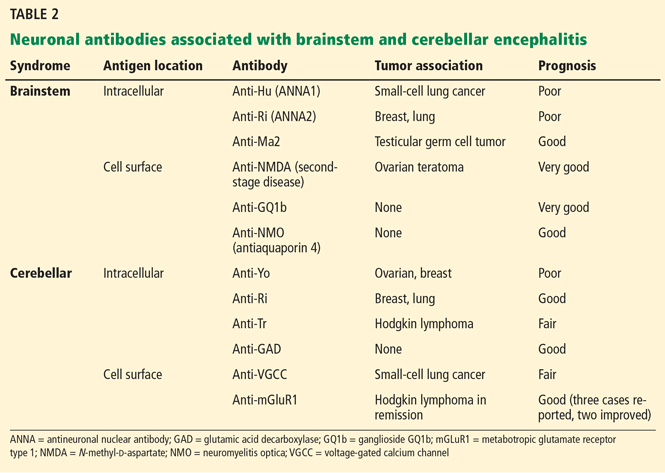
Anti-Hu, anti-Ri, and anti-Ma2 antibodies are most commonly associated with brainstem encephalitis (Table 2). Anti-Ma2-associated encephalitis may improve after a combination of immunotherapy and tumor removal21; the others have a poor prognosis.
Neuromyelitis optica spectrum disorders
Neuromyelitis optica spectrum disorders most commonly involve demyelination affecting the optic nerves and spinal cord, leading to unilateral or bilateral optic neuritis and transverse myelitis spanning three or more vertebral segments.22 The initial clinical manifestation may be an encephalitic pattern, affecting predominantly the brainstem in a restricted fashion,22 or the central nervous system in a more diffuse pattern, mimicking either acute disseminated encephalomyelitis or, in less severe cases, posterior reversible encephalopathy syndrome.23
Testing for antiaquaporin-4 antibody, also known as neuromyelitis optica immunoglobulin G, is the single most decisive laboratory test for diagnosing neuromyelitis optica spectrum disorders, so serum and cerebrospinal fluid evaluation for this autoantibody should be considered when caring for a patient whose clinical picture suggests brainstem encephalitis.22
Bickerstaff brainstem encephalitis
Bickerstaff brainstem encephalitis was first described more than half a century ago in patients with postinfectious ataxia, ophthalmoparesis, and altered consciousness. This rare disease was later found to be associated with antiganglioside GQ1b (anti-GQ1b) autoantibody. MRI is normal in about 90% of cases, so recognizing the clinical presentation and analyzing anti-GQ1b serum titers are critical to diagnosis.
Recovery is usually spontaneous and complete and can be hastened by immunotherapy, especially intravenous immunoglobulin.24
Other causes of brainstem encephalitis
The differential diagnosis of a presentation of brainstem encephalitis includes:
- Infectious causes, the most common being Listeria species followed by enterovirus 71 and herpes simplex virus.25 Tuberculosis, brucellosis, and Whipple disease should also be considered.
- Primary central nervous system inflammatory and demyelinating conditions, eg, multiple sclerosis and acute disseminated encephalomyelitis.
- Systemic inflammatory conditions, eg, Behçet disease, systemic lupus erythematosus, and sarcoidosis.
- Direct brainstem neoplastic involvement, as might occur in primary central nervous system lymphoma or leptomeningeal carcinomatosis.
CEREBELLAR SYNDROME
Patients with autoantibody-mediated encephalitis localized predominantly to the cerebellum typically present with dizziness, vertigo, and unsteady gait, progressing eventually to limb and gait ataxia.4 Symptoms are often subacute, progressing over weeks.
Multiple neuronal autoantibodies have been found to occur with cerebellar encephalitis (Table 2). In most cases, they are paraneoplastic and considered not to be pathogenic, given the intracellular location of their target antigen.4 In such cases, the syndrome is more accurately described as autoantibody-associated rather than autoantibody-mediated. Only in a minority of cases have neuronal autoantibodies been demonstrated to be directly pathogenic, ie, antimetabotropic glutamate receptor type 1 (anti-mGluR1) antibody-associated cerebellitis26 and antiglutamic acid decarboxylase (anti-GAD)-associated cerebellar ataxia.27
Differential diagnosis of cerebellar syndromes
The differential diagnosis of autoantibody-associated cerebellar syndromes is broad and includes:
- Alcohol-induced atrophy
- Drug-induced cerebellar atrophy (eg, from lithium, phenytoin, gabapentin, metronidazole, amiodarone, carbamazepine)
- Vitamin B1 and E deficiency
- Hypothyroidism, hypoparathyroidism
- Neurodegenerative disease (eg, prion disease, multiple system atrophy)
- Parainfectious causes (eg, after infection with Epstein-Barr virus)
- Immune-mediated diseases (Miller-Fisher syndrome, associated with anti-GQ1b antibodies, and antigliadin-associated ataxia, which can occur in isolation or as part of celiac disease).4
SYNDROMES ASSOCIATED WITH SPECIFIC ANTIBODIES
A few of the autoantibody-mediated encephalitic syndromes have specific antibody associations and characteristic clinical presentations. The most prominent of these syndromes are VGKC complex antibody encephalitis (as in the patient described at the beginning of this article) and anti-NMDA receptor encephalitis.
VGKC COMPLEX ANTIBODY-MEDIATED LIMBIC ENCEPHALITIS
VGKC complex antibodies, initially reported to be associated with the peripheral nerve hyperexcitability disorder neuromyotonia, were subsequently found in Morvan syndrome.28,29 Patients with this syndrome often present with autonomic dysfunction and peripheral nerve hyperexcitability but also develop insomnia, confusion, hallucinations, and memory loss. Drawing on the clinical overlap between Morvan syndrome and limbic encephalitis, Buckley et al30 were the first to report VGKC complex antibodies in two cases of limbic encephalitis.
VGKC complex antibodies are now understood to be associated with a wide variety of neurologic conditions, including chronic idiopathic pain, epilepsy,31 movement disorders, cranial nerve abnormalities, autonomic dysfunction,32 and gut dysmotility.33 In contrast, these antibodies are rare in healthy people.34 Limbic encephalitis associated with VGKC complex antibody usually lacks cerebellar and brainstem dysfunction, which may help distinguish it from other types of autoantibody-mediated limbic encephalitis.12
VGKC complex antibody does not bind to the potassium channel itself. Instead it recognizes other constituents of the channel complex, most notably LGI1 and contactin-associated protein 2 (CASPR2). LGI1 antibody is more commonly associated with limbic encephalitis—as illustrated in our case study—in addition to a distinctive type of seizure affecting the arm and face (faciobrachial dystonic seizure).34 The CASPR2 antibody, on the other hand, more often correlates with peripheral nerve manifestations and Morvan syndrome.29 Hyponatremia is commonly seen on serum chemical analysis and provides a clue that these syndromes are present.12
Good response to immunotherapy
A critical change in therapy came as clinicians realized that seizures were often refractory to standard antiepileptic drugs but responded well to immunotherapies. On the basis of these observations, sera of patients with long-standing epilepsy have been reanalyzed to look for neuronal autoantibodies.31 These antibodies should be checked in cases of new-onset refractory status epilepticus of unknown origin that does not respond to antiepileptic medications.
About half of patients with VGKC complex antibody-mediated limbic encephalitis have normal findings on brain MRI.5 Seven of 10 patients who were prospectively followed for VGKC complex antibody-mediated faciobrachial dystonic seizures had normal brain MRIs.35
VGKC complex antibody-mediated limbic encephalitis does not usually recur.36 Most cases are nonparaneoplastic, as evidenced by failure to detect a single active tumor in 64 patients after a median follow-up of 3 years. The prognosis is generally favorable except in cases with coexisting tumors.12
ANTI-NMDA RECEPTOR ENCEPHALITIS
Often associated with ovarian teratoma
Anti-NMDA receptor encephalitis typically affects women in their 20s and 30s, and about half of patients have an ovarian teratoma. It can also occur in younger patients and in men, in whom it is less likely to be associated with a neoplasm.37
Typical initial symptoms include striking and often stereotyped neuropsychiatric disturbances manifesting as psychosis, confusion, seizures, and amnesia. After 1 to 2 weeks, new symptoms set in, including reduced consciousness, movement disorders (ranging from orolingualfacial dyskinesia to rigidity and choreoathetosis), autonomic dysfunction, and hypoventilation, often prompting admission to the intensive care unit.38
Although the outcome is favorable in most cases, recovery, in contrast to VGKC complex antibody-mediated limbic encephalitis, is slow and may take longer than 1 year. Up to a quarter of patients have a relapse, underscoring the importance of maintenance immunotherapy.
It is important to undertake an intensive search for possible ovarian and extraovarian teratomas in young women with this syndrome—including CT of the pelvis, vaginal ultrasonography, and PET imaging—as removal of the teratoma may be curative.37
DIAGNOSIS OF AUTOANTIBODY-MEDIATED ENCEPHALITIS
Critical to diagnosing autoantibody-mediated encephalitis is awareness of these disorders. Since antibody testing may be very specific and is not usually part of the standard batteries of tests, a high level of suspicion is needed. Patients may present to different specialists in different settings; therefore, clinicians in pediatrics, rheumatology, psychiatry, and intensive care medicine need to be aware of these syndromes to avoid delay and misdiagnosis.
Clinical features suggesting autoantibody-mediated encephalitis include:
- Acute or subacute onset of a neurologic syndrome
- New-onset refractory status epilepticus of unknown etiology
- Acute or subacute psychiatric illness with unexpected progression to neurologic symptoms or delirium
- Unusual movement disorders not conforming to standard syndromes
- Cognitive impairment, psychosis, or behavioral or language disorders with atypical findings on imaging or cerebrospinal fluid analysis.
Imaging. Diagnosis of autoantibody-mediated encephalitis focuses on evidence suggesting an inflammatory central nervous system syndrome. MRI may show hyperintense signals on T2, FLAIR, or diffusion-weighted imaging changes in various brain regions. In many cases, however, MRI is negative despite severe clinical symptoms. In a study of 72 patients suspected of having autoimmune dementia of various etiologies, including but not restricted to antineuronal surface antibody-mediated causes, Flanagan et al39 identified atypical neuroimaging findings in only 29%. PET imaging may show hypermetabolism in certain brain areas correlating to clinical syndromes but is often difficult to obtain in a timely fashion.
Cerebrospinal fluid is often abnormal, showing elevated protein, increased immunoglobulin G synthesis, or oligoclonal banding. As with imaging studies, the cerebrospinal fluid may be normal despite severe clinical manifestations.
Electroencephalography may show focal slowing or seizure activity. Neuropsychologic testing may show different patterns of abnormalities.
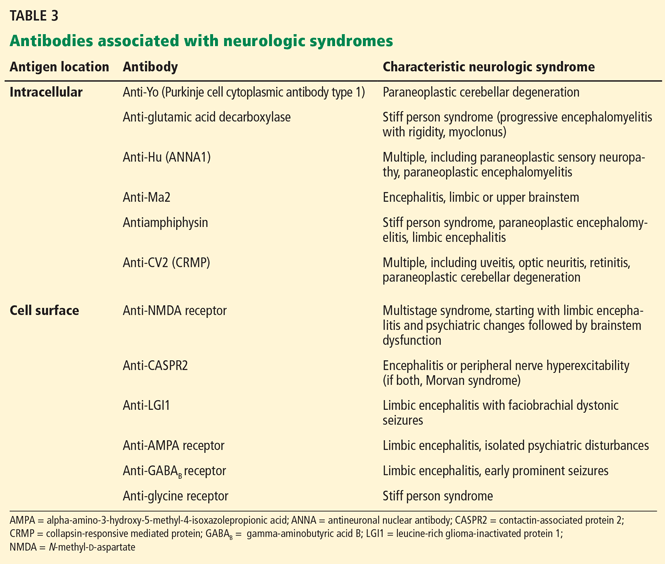
Antibody testing. None of these tests can be used in isolation, and the diagnosis of autoantibody-mediated encephalitis hinges on recognizing a clinical syndrome and ordering supportive testing. Specific antibodies are more likely in different clinical syndromes and should be sought (Table 3).
Patients who have autoantibody-mediated encephalitis may test negative for autoantibodies for many possible reasons:
- Blood testing for antibodies may be less sensitive than cerebrospinal fluid testing
- Antibody titers may vary in the course of the disease
- The patient may be expressing an antibody that is less often tested for (eg, anti-AMPA receptor or antigamma-aminobutyric acid B) or one that has not yet been isolated.
Evaluating for malignancy is recommended in all cases of autoantibody-mediated encephalitis. The initial workup may involve CT of the chest, abdomen, and pelvis, as well as mammography in women and serum prostate-specific antigen testing and testicular ultrasonography in men. Ordering FDG-PET in cases in which CT is negative or inconclusive increases cancer detection.40 If no cancer is found, close tumor surveillance—every 3 to 6 months—is recommended for at least 2 years.41
TREATMENT
Owing in large part to the rarity of autoantibody-mediated encephalitides, no randomized trials of therapy have been performed. Treatment at present is guided mostly by case series and expert consensus, which suggest first-line therapy with intravenous immunoglobulin, high-dose corticosteroids, plasmapheresis, or a combination.
Different syndromes and antibody-related disorders respond differently to therapy. Syndromes associated with antibodies against intracellular antigens tend to be more resistant to immune therapy than cell surface antigen-related syndromes.4
Tiered approach
Combined treatment with intravenous immunoglobulin and high-dose corticosteroids may be superior to treatment with steroids alone for LGI1-antibody mediated limbic encephalitis.42
In cases refractory to first-line (“tier 1”) therapy, second-line immunotherapy with drugs affecting B-cell populations (eg, rituximab, cyclophosphamide, and mycophenolate mofetil) has been used.
A tiered approach has been most extensively studied for anti-NMDA-receptor encephalitis, with better outcomes found using second-line therapy.43
Treatment strategies for these disorders will likely evolve over time with additional experience.
Outpatient management
Once the patient is discharged from the hospital, a multidisciplinary approach to care is recommended, including physical rehabilitation, speech therapy, neuropsychiatric and neuroimmunologic follow-up, and annual surveillance for malignancies.
- Brierley JB, Corsellis JAN, Hierons R, Nevin S. Subacute encephalitis of later adult life mainly affecting the limbic areas. Brain 1960; 83:357–368.
- Corsellis JA, Goldberg GJ, Norton AR. “Limbic encephalitis” and its association with carcinoma. Brain 1968; 91:481–496.
- Greenlee JE, Brashear HR. Antibodies to cerebellar Purkinje cells in patients with paraneoplastic cerebellar degeneration and ovarian carcinoma. Ann Neurol 1983; 14:609–613.
- Rosenfeld MR, Dalmau JO. Paraneoplastic disorders of the CNS and autoimmune synaptic encephalitis. Continuum (Minneap Minn) 2012; 18:366–383.
- Irani SR, Gelfand JM, Al-Diwani A, Vincent A. Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Ann Neurol 2014; 76:168–184.
- Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007; 61:25–36.
- Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 2010; 133:2734–2748.
- Lancaster E, Lai M, Peng X, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010; 9:67–76.
- Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009; 65:424–434.
- Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry 2012; 83:638–645.
- Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain 2005; 128:1764–1777.
- Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004; 127:701–712.
- Jammoul A, Lederman RJ, Tavee J, Li Y. Presence of voltage-gated potassium channel complex antibody in a case of genetic prion disease. BMJ Case Rep 2014; pii:bcr2013201622.
- Angus-Leppan H, Rudge P, Mead S, Collinge J, Vincent A. Autoantibodies in sporadic Creutzfeldt-Jakob disease. JAMA Neurol 2013; 70:919–922.
- Fujita K, Yuasa T, Watanabe O, et al. Voltage-gated potassium channel complex antibodies in Creutzfeldt-Jakob disease. J Neurol 2012; 259:2249–2250.
- Fujita K, Yuasa T, Takahashi Y, et al. Antibodies to N-methyl-D-aspartate glutamate receptors in Creutzfeldt–Jakob disease patients. J Neuroimmunol 2012; 251:90–93.
- Mackay G, Ahmad K, Stone J, et al. NMDA receptor autoantibodies in sporadic Creutzfeldt-Jakob disease. J Neurol 2012; 259:1979–1981.
- Leypoldt F, Titulaer MJ, Aguilar E, et al. Herpes simplex virus–1 encephalitis can trigger anti-NMDA receptor encephalitis: case report. Neurology 2013; 81:1637–1639.
- Desena A, Graves D, Warnack W, Greenberg BM. Herpes simplex encephalitis as a potential cause of anti-N-methyl-D-aspartate receptor antibody encephalitis: report of 2 cases. JAMA Neurol 2014; 71:344–346.
- Armangue T, Leypoldt F, Málaga I, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol 2014; 75:317–323.
- Blaes F. Paraneoplastic brain stem encephalitis. Curr Treat Options Neurol 2013; 15:201–209.
- Wildemann B, Jarius S. The expanding range of autoimmune disorders of the nervous system. Lancet Neurol 2013; 12:22–24.
- Kim W, Kim SH, Lee SH, Li XF, Kim HJ. Brain abnormalities as an initial manifestation of neuromyelitis optica spectrum disorder. Mult Scler 2011; 17:1107–1112.
- Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry 2013; 84:576–583.
- Jubelt B, Mihai C, Li MT, Veerapaneni P. Rhombencephalitis/brainstem encephalitis. Curr Neurol Neurosci Rep 2011; 11:543–552.
- Sillevis Smitt P, Kinoshita A, De Leeuw B, et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med 2000; 342:21–27.
- Ishida K, Mitoma H, Son SY, et al. Selective suppression of cerebellar GABAergic transmission by an autoantibody to glutamic acid decarboxylase. Ann Neurol 1999; 46:263–267.
- Hart IK, Waters C, Vincent A, et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol 1997; 41:238–246.
- Barber P, Anderson NE, Vincent A. Morvan’s syndrome associated with voltage-gated K+ channel antibodies. Neurology 2000; 54:771–772.
- Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001; 50:73–78.
- Majoie HJ, de Baets M, Renier W, Lang B, Vincent A. Antibodies to voltage-gated potassium and calcium channels in epilepsy. Epilepsy Res 2006; 71:135–141.
- Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology 2008; 70:1883–1890.
- Knowles CH, Lang B, Clover L, et al. A role for autoantibodies in some cases of acquired non-paraneoplastic gut dysmotility. Scand J Gastroenterol 2002; 37:166–170.
- Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011; 69:892–900.
- Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 2013: 136:3151–3162.
- Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol 2011; 10:759–772.
- Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011; 10:63–74.
- Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010; 133:1655–1667.
- Flanagan EP, McKeon A, Lennon VA, et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc 2010; 85:881–897.
- Younes-Mhenni S, Janier MF, Cinotti L, et al. FDG-PET improves tumour detection in patients with paraneoplastic neurological syndromes. Brain 2004; 127:2331–2338.
- Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011; 77:179–189.
- Shin YW, Lee ST, Shin JW, et al. VGKC-complex/LGI1-antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol 2013; 265:75–81.
- Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013; 12:157–165.
A 79-year-old woman with a history of breast cancer in remission and hypertension presented to a local emergency department because of subacute memory loss and compulsive shopping. Her serum sodium concentration was 127 mmol/L (reference range 132–148). Computed tomography (CT) and magnetic resonance imaging (MRI) of the brain were normal, and she was sent home.
Three days later, she experienced a generalized tonic-clonic seizure that evolved into status epilepticus. She was intubated and admitted to the intensive care unit. Cerebrospinal fluid analysis was normal, and infectious causes of encephalitis were ruled out. MRI showed increased signal in both hippocampi (Figure 1). Her seizures were refractory to treatment, and she was given pentobarbital to induce a coma.
Serum evaluation of neuronal antibodies revealed elevated titers of the voltage-gated potassium channel (VGKC) complex antibody, with subsequent subtyping confirming the leucine-rich glioma-inactivated protein 1 (LGI1) protein as the antigenic target.
She received a 5-day course of intravenous immunoglobulin and methylprednisolone, pentobarbital was withdrawn, and the seizures did not recur, but weeks later she remained comatose. Positron emission tomography (PET) of the brain revealed hypermetabolism in the medial and anterior aspects of both temporal lobes. She underwent five sessions of plasma exchange, after which she began to improve and follow commands. She was ultimately discharged to an acute rehabilitation facility after a 4-week hospital stay.
She received infusions of intravenous immunoglobulin twice a month for 6 months. At her last follow-up visit, she was seizure-free and neurologically intact except for mild inattention.
NEWLY RECOGNIZED DISEASES
Although autoantibody-mediated encephalitic syndromes were first described more than 50 years ago,1,2 their autoimmune basis was not recognized until the early 1980s.3 In the past 10 years, a flood of novel clinical syndromes associated with neuronal autoantibodies has been described that may be markedly improved or even completely resolved with immunotherapy. In cases of unexplained seizure, encephalitis, or acute-onset psychiatric syndromes, suspecting these syndromes can lead to diagnosis, treatment, and a good outcome.
This review describes the key clinical autoantibody-mediated encephalitic syndromes, explains the better-characterized antibody associations, and discusses their diagnosis and treatment.
CLASSIFIED ANATOMICALLY, IMMUNOLOGICALLY, OR EPONYMOUSLY
Autoantibody-mediated encephalitis is also known as autoimmune-mediated encephalitis, autoimmune-mediated limbic encephalitis, and autoimmune synaptic encephalitis.
How to categorize these syndromes is still in flux: they can be listed by the area of the brain affected, the antibody involved, or the name of the discoverer (eg, Morvan syndrome).
Autoantibodies identified in autoimmune encephalitis fall under two broad categories:
- Those targeting intracellular (intranuclear or intracytoplasmic) antigens; the syndromes they cause are more likely to be paraneoplastic and less responsive to immunotherapy
- Those targeting antigens on the neuronal surface: the syndromes they cause are less likely to be paraneoplastic and are more responsive to immunotherapy.4
SYNDROMES DEFINED BY BRAIN AREA AFFECTED
Below, we provide examples of neurologic syndromes of autoantibody-mediated encephalitis according to the region of the brain most affected, ie, the limbic system, the brainstem, or the cerebellum (Figure 2).
LIMBIC ENCEPHALITIS
Memory loss, behavioral changes, seizures
Patients with limbic encephalitis (such as the patient described in the vignette above) present with symptoms attributed to dysfunction of mesial temporal lobe structures, most notably the hippocampus. Prominent symptoms include short-term memory loss, behavioral disturbances such as agitation and confusion, and psychiatric problems such as depression and psychosis. Recurrent seizures are a salient feature and, not uncommonly, progress to status epilepticus.
Antibodies are not all cancer-associated
Cerebrospinal fluid analysis can be normal or show abnormalities suggesting immune activation, eg, slight pleocytosis, elevated protein, increased immunoglobulin G synthesis, and oligoclonal banding.5
In many cases, an autoantibody is found in the blood or in the cerebrospinal fluid. Some patients may express more than one autoantibody, so the traditional view of “one antibody, one syndrome” is incorrect.
Although initially identified as a rare paraneoplastic disorder, limbic encephalitis sometimes occurs in the absence of malignancy.
Multiple antibodies have been linked to the syndrome (Table 1).6–9 The “classic” antibodies initially found in paraneoplastic forms are now generally viewed as nonpathogenic, in part because they are directed against intracellular antigens. Neuronal injury in paraneoplastic limbic encephalitis is believed to be mediated by cytotoxic T lymphocytes, with neuronal autoantibodies being produced after the injury.4 Recently defined antibodies, such as those targeting the N-methyl-d-aspartate (NMDA) receptor6 and the LGI1 protein,7 are now understood to be common causes of limbic encephalitis.
Imaging usually shows limbic focal changes
Structural MRI or functional fluorodeoxyglucose (FDG)-PET imaging may show focal changes in limbic system structures, such as the mesial temporal lobes. It is now recognized that other cortical areas may be involved, and the term “limbic encephalitis” may give way to “cortical” or “focal encephalitis.”
In about 60% of patients, MRI shows hyperintense fluid-attenuated inversion recovery (FLAIR) or T2 signal changes in the mesial temporal lobes, likely reflecting inflammatory changes.4,10,11 On FDG-PET, hypermetabolism may be observed in the mesial temporal lobes early in the disease despite normal findings on MRI.12 Hypometabolism, either diffuse or localized to the mesial temporal lobes, eventually sets in, likely reflecting cytotoxic injury in the aftermath of prolonged inflammation or seizures.
Consider other causes
Before diagnosing limbic encephalitis, it is essential to evaluate for infectious meningoencephalitis, especially herpes simplex viral encephalitis. Thiamine deficiency (Wernicke encephalopathy), drug intoxication, prion disease, Hashimoto encephalopathy, tumor, and subclinical status epilepticus should also be considered. Some of these conditions are associated with the same neuronal autoantibodies detected in limbic encephalitis. Further complicating the picture, case reports have shown the presence of serum neuronal autoantibodies—VGKC complex13–15 and NMDA-receptor antibodies16,17—in confirmed cases of prion disease. In addition, adequately treated herpes simplex viral encephalitis can precipitate the production of NMDA-receptor antibodies and their characteristic syndrome.18–20
BRAINSTEM ENCEPHALITIS
The brainstem—the midbrain, pons, and medulla—can be affected, either in isolation or more commonly as part of a more widespread autoantibody-mediated encephalitis. Symptoms and signs include eye movement abnormalities, ptosis, dysphagia, dysarthria, ataxia, facial palsy, vertigo, hearing impairment, reduced consciousness, and hypoventilation.21
Anti-Hu, anti-Ri, and anti-Ma2 antibodies are most commonly associated with brainstem encephalitis (Table 2). Anti-Ma2-associated encephalitis may improve after a combination of immunotherapy and tumor removal21; the others have a poor prognosis.
Neuromyelitis optica spectrum disorders
Neuromyelitis optica spectrum disorders most commonly involve demyelination affecting the optic nerves and spinal cord, leading to unilateral or bilateral optic neuritis and transverse myelitis spanning three or more vertebral segments.22 The initial clinical manifestation may be an encephalitic pattern, affecting predominantly the brainstem in a restricted fashion,22 or the central nervous system in a more diffuse pattern, mimicking either acute disseminated encephalomyelitis or, in less severe cases, posterior reversible encephalopathy syndrome.23
Testing for antiaquaporin-4 antibody, also known as neuromyelitis optica immunoglobulin G, is the single most decisive laboratory test for diagnosing neuromyelitis optica spectrum disorders, so serum and cerebrospinal fluid evaluation for this autoantibody should be considered when caring for a patient whose clinical picture suggests brainstem encephalitis.22
Bickerstaff brainstem encephalitis
Bickerstaff brainstem encephalitis was first described more than half a century ago in patients with postinfectious ataxia, ophthalmoparesis, and altered consciousness. This rare disease was later found to be associated with antiganglioside GQ1b (anti-GQ1b) autoantibody. MRI is normal in about 90% of cases, so recognizing the clinical presentation and analyzing anti-GQ1b serum titers are critical to diagnosis.
Recovery is usually spontaneous and complete and can be hastened by immunotherapy, especially intravenous immunoglobulin.24
Other causes of brainstem encephalitis
The differential diagnosis of a presentation of brainstem encephalitis includes:
- Infectious causes, the most common being Listeria species followed by enterovirus 71 and herpes simplex virus.25 Tuberculosis, brucellosis, and Whipple disease should also be considered.
- Primary central nervous system inflammatory and demyelinating conditions, eg, multiple sclerosis and acute disseminated encephalomyelitis.
- Systemic inflammatory conditions, eg, Behçet disease, systemic lupus erythematosus, and sarcoidosis.
- Direct brainstem neoplastic involvement, as might occur in primary central nervous system lymphoma or leptomeningeal carcinomatosis.
CEREBELLAR SYNDROME
Patients with autoantibody-mediated encephalitis localized predominantly to the cerebellum typically present with dizziness, vertigo, and unsteady gait, progressing eventually to limb and gait ataxia.4 Symptoms are often subacute, progressing over weeks.
Multiple neuronal autoantibodies have been found to occur with cerebellar encephalitis (Table 2). In most cases, they are paraneoplastic and considered not to be pathogenic, given the intracellular location of their target antigen.4 In such cases, the syndrome is more accurately described as autoantibody-associated rather than autoantibody-mediated. Only in a minority of cases have neuronal autoantibodies been demonstrated to be directly pathogenic, ie, antimetabotropic glutamate receptor type 1 (anti-mGluR1) antibody-associated cerebellitis26 and antiglutamic acid decarboxylase (anti-GAD)-associated cerebellar ataxia.27
Differential diagnosis of cerebellar syndromes
The differential diagnosis of autoantibody-associated cerebellar syndromes is broad and includes:
- Alcohol-induced atrophy
- Drug-induced cerebellar atrophy (eg, from lithium, phenytoin, gabapentin, metronidazole, amiodarone, carbamazepine)
- Vitamin B1 and E deficiency
- Hypothyroidism, hypoparathyroidism
- Neurodegenerative disease (eg, prion disease, multiple system atrophy)
- Parainfectious causes (eg, after infection with Epstein-Barr virus)
- Immune-mediated diseases (Miller-Fisher syndrome, associated with anti-GQ1b antibodies, and antigliadin-associated ataxia, which can occur in isolation or as part of celiac disease).4
SYNDROMES ASSOCIATED WITH SPECIFIC ANTIBODIES
A few of the autoantibody-mediated encephalitic syndromes have specific antibody associations and characteristic clinical presentations. The most prominent of these syndromes are VGKC complex antibody encephalitis (as in the patient described at the beginning of this article) and anti-NMDA receptor encephalitis.
VGKC COMPLEX ANTIBODY-MEDIATED LIMBIC ENCEPHALITIS
VGKC complex antibodies, initially reported to be associated with the peripheral nerve hyperexcitability disorder neuromyotonia, were subsequently found in Morvan syndrome.28,29 Patients with this syndrome often present with autonomic dysfunction and peripheral nerve hyperexcitability but also develop insomnia, confusion, hallucinations, and memory loss. Drawing on the clinical overlap between Morvan syndrome and limbic encephalitis, Buckley et al30 were the first to report VGKC complex antibodies in two cases of limbic encephalitis.
VGKC complex antibodies are now understood to be associated with a wide variety of neurologic conditions, including chronic idiopathic pain, epilepsy,31 movement disorders, cranial nerve abnormalities, autonomic dysfunction,32 and gut dysmotility.33 In contrast, these antibodies are rare in healthy people.34 Limbic encephalitis associated with VGKC complex antibody usually lacks cerebellar and brainstem dysfunction, which may help distinguish it from other types of autoantibody-mediated limbic encephalitis.12
VGKC complex antibody does not bind to the potassium channel itself. Instead it recognizes other constituents of the channel complex, most notably LGI1 and contactin-associated protein 2 (CASPR2). LGI1 antibody is more commonly associated with limbic encephalitis—as illustrated in our case study—in addition to a distinctive type of seizure affecting the arm and face (faciobrachial dystonic seizure).34 The CASPR2 antibody, on the other hand, more often correlates with peripheral nerve manifestations and Morvan syndrome.29 Hyponatremia is commonly seen on serum chemical analysis and provides a clue that these syndromes are present.12
Good response to immunotherapy
A critical change in therapy came as clinicians realized that seizures were often refractory to standard antiepileptic drugs but responded well to immunotherapies. On the basis of these observations, sera of patients with long-standing epilepsy have been reanalyzed to look for neuronal autoantibodies.31 These antibodies should be checked in cases of new-onset refractory status epilepticus of unknown origin that does not respond to antiepileptic medications.
About half of patients with VGKC complex antibody-mediated limbic encephalitis have normal findings on brain MRI.5 Seven of 10 patients who were prospectively followed for VGKC complex antibody-mediated faciobrachial dystonic seizures had normal brain MRIs.35
VGKC complex antibody-mediated limbic encephalitis does not usually recur.36 Most cases are nonparaneoplastic, as evidenced by failure to detect a single active tumor in 64 patients after a median follow-up of 3 years. The prognosis is generally favorable except in cases with coexisting tumors.12
ANTI-NMDA RECEPTOR ENCEPHALITIS
Often associated with ovarian teratoma
Anti-NMDA receptor encephalitis typically affects women in their 20s and 30s, and about half of patients have an ovarian teratoma. It can also occur in younger patients and in men, in whom it is less likely to be associated with a neoplasm.37
Typical initial symptoms include striking and often stereotyped neuropsychiatric disturbances manifesting as psychosis, confusion, seizures, and amnesia. After 1 to 2 weeks, new symptoms set in, including reduced consciousness, movement disorders (ranging from orolingualfacial dyskinesia to rigidity and choreoathetosis), autonomic dysfunction, and hypoventilation, often prompting admission to the intensive care unit.38
Although the outcome is favorable in most cases, recovery, in contrast to VGKC complex antibody-mediated limbic encephalitis, is slow and may take longer than 1 year. Up to a quarter of patients have a relapse, underscoring the importance of maintenance immunotherapy.
It is important to undertake an intensive search for possible ovarian and extraovarian teratomas in young women with this syndrome—including CT of the pelvis, vaginal ultrasonography, and PET imaging—as removal of the teratoma may be curative.37
DIAGNOSIS OF AUTOANTIBODY-MEDIATED ENCEPHALITIS
Critical to diagnosing autoantibody-mediated encephalitis is awareness of these disorders. Since antibody testing may be very specific and is not usually part of the standard batteries of tests, a high level of suspicion is needed. Patients may present to different specialists in different settings; therefore, clinicians in pediatrics, rheumatology, psychiatry, and intensive care medicine need to be aware of these syndromes to avoid delay and misdiagnosis.
Clinical features suggesting autoantibody-mediated encephalitis include:
- Acute or subacute onset of a neurologic syndrome
- New-onset refractory status epilepticus of unknown etiology
- Acute or subacute psychiatric illness with unexpected progression to neurologic symptoms or delirium
- Unusual movement disorders not conforming to standard syndromes
- Cognitive impairment, psychosis, or behavioral or language disorders with atypical findings on imaging or cerebrospinal fluid analysis.
Imaging. Diagnosis of autoantibody-mediated encephalitis focuses on evidence suggesting an inflammatory central nervous system syndrome. MRI may show hyperintense signals on T2, FLAIR, or diffusion-weighted imaging changes in various brain regions. In many cases, however, MRI is negative despite severe clinical symptoms. In a study of 72 patients suspected of having autoimmune dementia of various etiologies, including but not restricted to antineuronal surface antibody-mediated causes, Flanagan et al39 identified atypical neuroimaging findings in only 29%. PET imaging may show hypermetabolism in certain brain areas correlating to clinical syndromes but is often difficult to obtain in a timely fashion.
Cerebrospinal fluid is often abnormal, showing elevated protein, increased immunoglobulin G synthesis, or oligoclonal banding. As with imaging studies, the cerebrospinal fluid may be normal despite severe clinical manifestations.
Electroencephalography may show focal slowing or seizure activity. Neuropsychologic testing may show different patterns of abnormalities.
Antibody testing. None of these tests can be used in isolation, and the diagnosis of autoantibody-mediated encephalitis hinges on recognizing a clinical syndrome and ordering supportive testing. Specific antibodies are more likely in different clinical syndromes and should be sought (Table 3).
Patients who have autoantibody-mediated encephalitis may test negative for autoantibodies for many possible reasons:
- Blood testing for antibodies may be less sensitive than cerebrospinal fluid testing
- Antibody titers may vary in the course of the disease
- The patient may be expressing an antibody that is less often tested for (eg, anti-AMPA receptor or antigamma-aminobutyric acid B) or one that has not yet been isolated.
Evaluating for malignancy is recommended in all cases of autoantibody-mediated encephalitis. The initial workup may involve CT of the chest, abdomen, and pelvis, as well as mammography in women and serum prostate-specific antigen testing and testicular ultrasonography in men. Ordering FDG-PET in cases in which CT is negative or inconclusive increases cancer detection.40 If no cancer is found, close tumor surveillance—every 3 to 6 months—is recommended for at least 2 years.41
TREATMENT
Owing in large part to the rarity of autoantibody-mediated encephalitides, no randomized trials of therapy have been performed. Treatment at present is guided mostly by case series and expert consensus, which suggest first-line therapy with intravenous immunoglobulin, high-dose corticosteroids, plasmapheresis, or a combination.
Different syndromes and antibody-related disorders respond differently to therapy. Syndromes associated with antibodies against intracellular antigens tend to be more resistant to immune therapy than cell surface antigen-related syndromes.4
Tiered approach
Combined treatment with intravenous immunoglobulin and high-dose corticosteroids may be superior to treatment with steroids alone for LGI1-antibody mediated limbic encephalitis.42
In cases refractory to first-line (“tier 1”) therapy, second-line immunotherapy with drugs affecting B-cell populations (eg, rituximab, cyclophosphamide, and mycophenolate mofetil) has been used.
A tiered approach has been most extensively studied for anti-NMDA-receptor encephalitis, with better outcomes found using second-line therapy.43
Treatment strategies for these disorders will likely evolve over time with additional experience.
Outpatient management
Once the patient is discharged from the hospital, a multidisciplinary approach to care is recommended, including physical rehabilitation, speech therapy, neuropsychiatric and neuroimmunologic follow-up, and annual surveillance for malignancies.
A 79-year-old woman with a history of breast cancer in remission and hypertension presented to a local emergency department because of subacute memory loss and compulsive shopping. Her serum sodium concentration was 127 mmol/L (reference range 132–148). Computed tomography (CT) and magnetic resonance imaging (MRI) of the brain were normal, and she was sent home.
Three days later, she experienced a generalized tonic-clonic seizure that evolved into status epilepticus. She was intubated and admitted to the intensive care unit. Cerebrospinal fluid analysis was normal, and infectious causes of encephalitis were ruled out. MRI showed increased signal in both hippocampi (Figure 1). Her seizures were refractory to treatment, and she was given pentobarbital to induce a coma.
Serum evaluation of neuronal antibodies revealed elevated titers of the voltage-gated potassium channel (VGKC) complex antibody, with subsequent subtyping confirming the leucine-rich glioma-inactivated protein 1 (LGI1) protein as the antigenic target.
She received a 5-day course of intravenous immunoglobulin and methylprednisolone, pentobarbital was withdrawn, and the seizures did not recur, but weeks later she remained comatose. Positron emission tomography (PET) of the brain revealed hypermetabolism in the medial and anterior aspects of both temporal lobes. She underwent five sessions of plasma exchange, after which she began to improve and follow commands. She was ultimately discharged to an acute rehabilitation facility after a 4-week hospital stay.
She received infusions of intravenous immunoglobulin twice a month for 6 months. At her last follow-up visit, she was seizure-free and neurologically intact except for mild inattention.
NEWLY RECOGNIZED DISEASES
Although autoantibody-mediated encephalitic syndromes were first described more than 50 years ago,1,2 their autoimmune basis was not recognized until the early 1980s.3 In the past 10 years, a flood of novel clinical syndromes associated with neuronal autoantibodies has been described that may be markedly improved or even completely resolved with immunotherapy. In cases of unexplained seizure, encephalitis, or acute-onset psychiatric syndromes, suspecting these syndromes can lead to diagnosis, treatment, and a good outcome.
This review describes the key clinical autoantibody-mediated encephalitic syndromes, explains the better-characterized antibody associations, and discusses their diagnosis and treatment.
CLASSIFIED ANATOMICALLY, IMMUNOLOGICALLY, OR EPONYMOUSLY
Autoantibody-mediated encephalitis is also known as autoimmune-mediated encephalitis, autoimmune-mediated limbic encephalitis, and autoimmune synaptic encephalitis.
How to categorize these syndromes is still in flux: they can be listed by the area of the brain affected, the antibody involved, or the name of the discoverer (eg, Morvan syndrome).
Autoantibodies identified in autoimmune encephalitis fall under two broad categories:
- Those targeting intracellular (intranuclear or intracytoplasmic) antigens; the syndromes they cause are more likely to be paraneoplastic and less responsive to immunotherapy
- Those targeting antigens on the neuronal surface: the syndromes they cause are less likely to be paraneoplastic and are more responsive to immunotherapy.4
SYNDROMES DEFINED BY BRAIN AREA AFFECTED
Below, we provide examples of neurologic syndromes of autoantibody-mediated encephalitis according to the region of the brain most affected, ie, the limbic system, the brainstem, or the cerebellum (Figure 2).
LIMBIC ENCEPHALITIS
Memory loss, behavioral changes, seizures
Patients with limbic encephalitis (such as the patient described in the vignette above) present with symptoms attributed to dysfunction of mesial temporal lobe structures, most notably the hippocampus. Prominent symptoms include short-term memory loss, behavioral disturbances such as agitation and confusion, and psychiatric problems such as depression and psychosis. Recurrent seizures are a salient feature and, not uncommonly, progress to status epilepticus.
Antibodies are not all cancer-associated
Cerebrospinal fluid analysis can be normal or show abnormalities suggesting immune activation, eg, slight pleocytosis, elevated protein, increased immunoglobulin G synthesis, and oligoclonal banding.5
In many cases, an autoantibody is found in the blood or in the cerebrospinal fluid. Some patients may express more than one autoantibody, so the traditional view of “one antibody, one syndrome” is incorrect.
Although initially identified as a rare paraneoplastic disorder, limbic encephalitis sometimes occurs in the absence of malignancy.
Multiple antibodies have been linked to the syndrome (Table 1).6–9 The “classic” antibodies initially found in paraneoplastic forms are now generally viewed as nonpathogenic, in part because they are directed against intracellular antigens. Neuronal injury in paraneoplastic limbic encephalitis is believed to be mediated by cytotoxic T lymphocytes, with neuronal autoantibodies being produced after the injury.4 Recently defined antibodies, such as those targeting the N-methyl-d-aspartate (NMDA) receptor6 and the LGI1 protein,7 are now understood to be common causes of limbic encephalitis.
Imaging usually shows limbic focal changes
Structural MRI or functional fluorodeoxyglucose (FDG)-PET imaging may show focal changes in limbic system structures, such as the mesial temporal lobes. It is now recognized that other cortical areas may be involved, and the term “limbic encephalitis” may give way to “cortical” or “focal encephalitis.”
In about 60% of patients, MRI shows hyperintense fluid-attenuated inversion recovery (FLAIR) or T2 signal changes in the mesial temporal lobes, likely reflecting inflammatory changes.4,10,11 On FDG-PET, hypermetabolism may be observed in the mesial temporal lobes early in the disease despite normal findings on MRI.12 Hypometabolism, either diffuse or localized to the mesial temporal lobes, eventually sets in, likely reflecting cytotoxic injury in the aftermath of prolonged inflammation or seizures.
Consider other causes
Before diagnosing limbic encephalitis, it is essential to evaluate for infectious meningoencephalitis, especially herpes simplex viral encephalitis. Thiamine deficiency (Wernicke encephalopathy), drug intoxication, prion disease, Hashimoto encephalopathy, tumor, and subclinical status epilepticus should also be considered. Some of these conditions are associated with the same neuronal autoantibodies detected in limbic encephalitis. Further complicating the picture, case reports have shown the presence of serum neuronal autoantibodies—VGKC complex13–15 and NMDA-receptor antibodies16,17—in confirmed cases of prion disease. In addition, adequately treated herpes simplex viral encephalitis can precipitate the production of NMDA-receptor antibodies and their characteristic syndrome.18–20
BRAINSTEM ENCEPHALITIS
The brainstem—the midbrain, pons, and medulla—can be affected, either in isolation or more commonly as part of a more widespread autoantibody-mediated encephalitis. Symptoms and signs include eye movement abnormalities, ptosis, dysphagia, dysarthria, ataxia, facial palsy, vertigo, hearing impairment, reduced consciousness, and hypoventilation.21
Anti-Hu, anti-Ri, and anti-Ma2 antibodies are most commonly associated with brainstem encephalitis (Table 2). Anti-Ma2-associated encephalitis may improve after a combination of immunotherapy and tumor removal21; the others have a poor prognosis.
Neuromyelitis optica spectrum disorders
Neuromyelitis optica spectrum disorders most commonly involve demyelination affecting the optic nerves and spinal cord, leading to unilateral or bilateral optic neuritis and transverse myelitis spanning three or more vertebral segments.22 The initial clinical manifestation may be an encephalitic pattern, affecting predominantly the brainstem in a restricted fashion,22 or the central nervous system in a more diffuse pattern, mimicking either acute disseminated encephalomyelitis or, in less severe cases, posterior reversible encephalopathy syndrome.23
Testing for antiaquaporin-4 antibody, also known as neuromyelitis optica immunoglobulin G, is the single most decisive laboratory test for diagnosing neuromyelitis optica spectrum disorders, so serum and cerebrospinal fluid evaluation for this autoantibody should be considered when caring for a patient whose clinical picture suggests brainstem encephalitis.22
Bickerstaff brainstem encephalitis
Bickerstaff brainstem encephalitis was first described more than half a century ago in patients with postinfectious ataxia, ophthalmoparesis, and altered consciousness. This rare disease was later found to be associated with antiganglioside GQ1b (anti-GQ1b) autoantibody. MRI is normal in about 90% of cases, so recognizing the clinical presentation and analyzing anti-GQ1b serum titers are critical to diagnosis.
Recovery is usually spontaneous and complete and can be hastened by immunotherapy, especially intravenous immunoglobulin.24
Other causes of brainstem encephalitis
The differential diagnosis of a presentation of brainstem encephalitis includes:
- Infectious causes, the most common being Listeria species followed by enterovirus 71 and herpes simplex virus.25 Tuberculosis, brucellosis, and Whipple disease should also be considered.
- Primary central nervous system inflammatory and demyelinating conditions, eg, multiple sclerosis and acute disseminated encephalomyelitis.
- Systemic inflammatory conditions, eg, Behçet disease, systemic lupus erythematosus, and sarcoidosis.
- Direct brainstem neoplastic involvement, as might occur in primary central nervous system lymphoma or leptomeningeal carcinomatosis.
CEREBELLAR SYNDROME
Patients with autoantibody-mediated encephalitis localized predominantly to the cerebellum typically present with dizziness, vertigo, and unsteady gait, progressing eventually to limb and gait ataxia.4 Symptoms are often subacute, progressing over weeks.
Multiple neuronal autoantibodies have been found to occur with cerebellar encephalitis (Table 2). In most cases, they are paraneoplastic and considered not to be pathogenic, given the intracellular location of their target antigen.4 In such cases, the syndrome is more accurately described as autoantibody-associated rather than autoantibody-mediated. Only in a minority of cases have neuronal autoantibodies been demonstrated to be directly pathogenic, ie, antimetabotropic glutamate receptor type 1 (anti-mGluR1) antibody-associated cerebellitis26 and antiglutamic acid decarboxylase (anti-GAD)-associated cerebellar ataxia.27
Differential diagnosis of cerebellar syndromes
The differential diagnosis of autoantibody-associated cerebellar syndromes is broad and includes:
- Alcohol-induced atrophy
- Drug-induced cerebellar atrophy (eg, from lithium, phenytoin, gabapentin, metronidazole, amiodarone, carbamazepine)
- Vitamin B1 and E deficiency
- Hypothyroidism, hypoparathyroidism
- Neurodegenerative disease (eg, prion disease, multiple system atrophy)
- Parainfectious causes (eg, after infection with Epstein-Barr virus)
- Immune-mediated diseases (Miller-Fisher syndrome, associated with anti-GQ1b antibodies, and antigliadin-associated ataxia, which can occur in isolation or as part of celiac disease).4
SYNDROMES ASSOCIATED WITH SPECIFIC ANTIBODIES
A few of the autoantibody-mediated encephalitic syndromes have specific antibody associations and characteristic clinical presentations. The most prominent of these syndromes are VGKC complex antibody encephalitis (as in the patient described at the beginning of this article) and anti-NMDA receptor encephalitis.
VGKC COMPLEX ANTIBODY-MEDIATED LIMBIC ENCEPHALITIS
VGKC complex antibodies, initially reported to be associated with the peripheral nerve hyperexcitability disorder neuromyotonia, were subsequently found in Morvan syndrome.28,29 Patients with this syndrome often present with autonomic dysfunction and peripheral nerve hyperexcitability but also develop insomnia, confusion, hallucinations, and memory loss. Drawing on the clinical overlap between Morvan syndrome and limbic encephalitis, Buckley et al30 were the first to report VGKC complex antibodies in two cases of limbic encephalitis.
VGKC complex antibodies are now understood to be associated with a wide variety of neurologic conditions, including chronic idiopathic pain, epilepsy,31 movement disorders, cranial nerve abnormalities, autonomic dysfunction,32 and gut dysmotility.33 In contrast, these antibodies are rare in healthy people.34 Limbic encephalitis associated with VGKC complex antibody usually lacks cerebellar and brainstem dysfunction, which may help distinguish it from other types of autoantibody-mediated limbic encephalitis.12
VGKC complex antibody does not bind to the potassium channel itself. Instead it recognizes other constituents of the channel complex, most notably LGI1 and contactin-associated protein 2 (CASPR2). LGI1 antibody is more commonly associated with limbic encephalitis—as illustrated in our case study—in addition to a distinctive type of seizure affecting the arm and face (faciobrachial dystonic seizure).34 The CASPR2 antibody, on the other hand, more often correlates with peripheral nerve manifestations and Morvan syndrome.29 Hyponatremia is commonly seen on serum chemical analysis and provides a clue that these syndromes are present.12
Good response to immunotherapy
A critical change in therapy came as clinicians realized that seizures were often refractory to standard antiepileptic drugs but responded well to immunotherapies. On the basis of these observations, sera of patients with long-standing epilepsy have been reanalyzed to look for neuronal autoantibodies.31 These antibodies should be checked in cases of new-onset refractory status epilepticus of unknown origin that does not respond to antiepileptic medications.
About half of patients with VGKC complex antibody-mediated limbic encephalitis have normal findings on brain MRI.5 Seven of 10 patients who were prospectively followed for VGKC complex antibody-mediated faciobrachial dystonic seizures had normal brain MRIs.35
VGKC complex antibody-mediated limbic encephalitis does not usually recur.36 Most cases are nonparaneoplastic, as evidenced by failure to detect a single active tumor in 64 patients after a median follow-up of 3 years. The prognosis is generally favorable except in cases with coexisting tumors.12
ANTI-NMDA RECEPTOR ENCEPHALITIS
Often associated with ovarian teratoma
Anti-NMDA receptor encephalitis typically affects women in their 20s and 30s, and about half of patients have an ovarian teratoma. It can also occur in younger patients and in men, in whom it is less likely to be associated with a neoplasm.37
Typical initial symptoms include striking and often stereotyped neuropsychiatric disturbances manifesting as psychosis, confusion, seizures, and amnesia. After 1 to 2 weeks, new symptoms set in, including reduced consciousness, movement disorders (ranging from orolingualfacial dyskinesia to rigidity and choreoathetosis), autonomic dysfunction, and hypoventilation, often prompting admission to the intensive care unit.38
Although the outcome is favorable in most cases, recovery, in contrast to VGKC complex antibody-mediated limbic encephalitis, is slow and may take longer than 1 year. Up to a quarter of patients have a relapse, underscoring the importance of maintenance immunotherapy.
It is important to undertake an intensive search for possible ovarian and extraovarian teratomas in young women with this syndrome—including CT of the pelvis, vaginal ultrasonography, and PET imaging—as removal of the teratoma may be curative.37
DIAGNOSIS OF AUTOANTIBODY-MEDIATED ENCEPHALITIS
Critical to diagnosing autoantibody-mediated encephalitis is awareness of these disorders. Since antibody testing may be very specific and is not usually part of the standard batteries of tests, a high level of suspicion is needed. Patients may present to different specialists in different settings; therefore, clinicians in pediatrics, rheumatology, psychiatry, and intensive care medicine need to be aware of these syndromes to avoid delay and misdiagnosis.
Clinical features suggesting autoantibody-mediated encephalitis include:
- Acute or subacute onset of a neurologic syndrome
- New-onset refractory status epilepticus of unknown etiology
- Acute or subacute psychiatric illness with unexpected progression to neurologic symptoms or delirium
- Unusual movement disorders not conforming to standard syndromes
- Cognitive impairment, psychosis, or behavioral or language disorders with atypical findings on imaging or cerebrospinal fluid analysis.
Imaging. Diagnosis of autoantibody-mediated encephalitis focuses on evidence suggesting an inflammatory central nervous system syndrome. MRI may show hyperintense signals on T2, FLAIR, or diffusion-weighted imaging changes in various brain regions. In many cases, however, MRI is negative despite severe clinical symptoms. In a study of 72 patients suspected of having autoimmune dementia of various etiologies, including but not restricted to antineuronal surface antibody-mediated causes, Flanagan et al39 identified atypical neuroimaging findings in only 29%. PET imaging may show hypermetabolism in certain brain areas correlating to clinical syndromes but is often difficult to obtain in a timely fashion.
Cerebrospinal fluid is often abnormal, showing elevated protein, increased immunoglobulin G synthesis, or oligoclonal banding. As with imaging studies, the cerebrospinal fluid may be normal despite severe clinical manifestations.
Electroencephalography may show focal slowing or seizure activity. Neuropsychologic testing may show different patterns of abnormalities.
Antibody testing. None of these tests can be used in isolation, and the diagnosis of autoantibody-mediated encephalitis hinges on recognizing a clinical syndrome and ordering supportive testing. Specific antibodies are more likely in different clinical syndromes and should be sought (Table 3).
Patients who have autoantibody-mediated encephalitis may test negative for autoantibodies for many possible reasons:
- Blood testing for antibodies may be less sensitive than cerebrospinal fluid testing
- Antibody titers may vary in the course of the disease
- The patient may be expressing an antibody that is less often tested for (eg, anti-AMPA receptor or antigamma-aminobutyric acid B) or one that has not yet been isolated.
Evaluating for malignancy is recommended in all cases of autoantibody-mediated encephalitis. The initial workup may involve CT of the chest, abdomen, and pelvis, as well as mammography in women and serum prostate-specific antigen testing and testicular ultrasonography in men. Ordering FDG-PET in cases in which CT is negative or inconclusive increases cancer detection.40 If no cancer is found, close tumor surveillance—every 3 to 6 months—is recommended for at least 2 years.41
TREATMENT
Owing in large part to the rarity of autoantibody-mediated encephalitides, no randomized trials of therapy have been performed. Treatment at present is guided mostly by case series and expert consensus, which suggest first-line therapy with intravenous immunoglobulin, high-dose corticosteroids, plasmapheresis, or a combination.
Different syndromes and antibody-related disorders respond differently to therapy. Syndromes associated with antibodies against intracellular antigens tend to be more resistant to immune therapy than cell surface antigen-related syndromes.4
Tiered approach
Combined treatment with intravenous immunoglobulin and high-dose corticosteroids may be superior to treatment with steroids alone for LGI1-antibody mediated limbic encephalitis.42
In cases refractory to first-line (“tier 1”) therapy, second-line immunotherapy with drugs affecting B-cell populations (eg, rituximab, cyclophosphamide, and mycophenolate mofetil) has been used.
A tiered approach has been most extensively studied for anti-NMDA-receptor encephalitis, with better outcomes found using second-line therapy.43
Treatment strategies for these disorders will likely evolve over time with additional experience.
Outpatient management
Once the patient is discharged from the hospital, a multidisciplinary approach to care is recommended, including physical rehabilitation, speech therapy, neuropsychiatric and neuroimmunologic follow-up, and annual surveillance for malignancies.
- Brierley JB, Corsellis JAN, Hierons R, Nevin S. Subacute encephalitis of later adult life mainly affecting the limbic areas. Brain 1960; 83:357–368.
- Corsellis JA, Goldberg GJ, Norton AR. “Limbic encephalitis” and its association with carcinoma. Brain 1968; 91:481–496.
- Greenlee JE, Brashear HR. Antibodies to cerebellar Purkinje cells in patients with paraneoplastic cerebellar degeneration and ovarian carcinoma. Ann Neurol 1983; 14:609–613.
- Rosenfeld MR, Dalmau JO. Paraneoplastic disorders of the CNS and autoimmune synaptic encephalitis. Continuum (Minneap Minn) 2012; 18:366–383.
- Irani SR, Gelfand JM, Al-Diwani A, Vincent A. Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Ann Neurol 2014; 76:168–184.
- Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007; 61:25–36.
- Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 2010; 133:2734–2748.
- Lancaster E, Lai M, Peng X, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010; 9:67–76.
- Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009; 65:424–434.
- Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry 2012; 83:638–645.
- Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain 2005; 128:1764–1777.
- Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004; 127:701–712.
- Jammoul A, Lederman RJ, Tavee J, Li Y. Presence of voltage-gated potassium channel complex antibody in a case of genetic prion disease. BMJ Case Rep 2014; pii:bcr2013201622.
- Angus-Leppan H, Rudge P, Mead S, Collinge J, Vincent A. Autoantibodies in sporadic Creutzfeldt-Jakob disease. JAMA Neurol 2013; 70:919–922.
- Fujita K, Yuasa T, Watanabe O, et al. Voltage-gated potassium channel complex antibodies in Creutzfeldt-Jakob disease. J Neurol 2012; 259:2249–2250.
- Fujita K, Yuasa T, Takahashi Y, et al. Antibodies to N-methyl-D-aspartate glutamate receptors in Creutzfeldt–Jakob disease patients. J Neuroimmunol 2012; 251:90–93.
- Mackay G, Ahmad K, Stone J, et al. NMDA receptor autoantibodies in sporadic Creutzfeldt-Jakob disease. J Neurol 2012; 259:1979–1981.
- Leypoldt F, Titulaer MJ, Aguilar E, et al. Herpes simplex virus–1 encephalitis can trigger anti-NMDA receptor encephalitis: case report. Neurology 2013; 81:1637–1639.
- Desena A, Graves D, Warnack W, Greenberg BM. Herpes simplex encephalitis as a potential cause of anti-N-methyl-D-aspartate receptor antibody encephalitis: report of 2 cases. JAMA Neurol 2014; 71:344–346.
- Armangue T, Leypoldt F, Málaga I, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol 2014; 75:317–323.
- Blaes F. Paraneoplastic brain stem encephalitis. Curr Treat Options Neurol 2013; 15:201–209.
- Wildemann B, Jarius S. The expanding range of autoimmune disorders of the nervous system. Lancet Neurol 2013; 12:22–24.
- Kim W, Kim SH, Lee SH, Li XF, Kim HJ. Brain abnormalities as an initial manifestation of neuromyelitis optica spectrum disorder. Mult Scler 2011; 17:1107–1112.
- Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry 2013; 84:576–583.
- Jubelt B, Mihai C, Li MT, Veerapaneni P. Rhombencephalitis/brainstem encephalitis. Curr Neurol Neurosci Rep 2011; 11:543–552.
- Sillevis Smitt P, Kinoshita A, De Leeuw B, et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med 2000; 342:21–27.
- Ishida K, Mitoma H, Son SY, et al. Selective suppression of cerebellar GABAergic transmission by an autoantibody to glutamic acid decarboxylase. Ann Neurol 1999; 46:263–267.
- Hart IK, Waters C, Vincent A, et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol 1997; 41:238–246.
- Barber P, Anderson NE, Vincent A. Morvan’s syndrome associated with voltage-gated K+ channel antibodies. Neurology 2000; 54:771–772.
- Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001; 50:73–78.
- Majoie HJ, de Baets M, Renier W, Lang B, Vincent A. Antibodies to voltage-gated potassium and calcium channels in epilepsy. Epilepsy Res 2006; 71:135–141.
- Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology 2008; 70:1883–1890.
- Knowles CH, Lang B, Clover L, et al. A role for autoantibodies in some cases of acquired non-paraneoplastic gut dysmotility. Scand J Gastroenterol 2002; 37:166–170.
- Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011; 69:892–900.
- Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 2013: 136:3151–3162.
- Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol 2011; 10:759–772.
- Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011; 10:63–74.
- Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010; 133:1655–1667.
- Flanagan EP, McKeon A, Lennon VA, et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc 2010; 85:881–897.
- Younes-Mhenni S, Janier MF, Cinotti L, et al. FDG-PET improves tumour detection in patients with paraneoplastic neurological syndromes. Brain 2004; 127:2331–2338.
- Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011; 77:179–189.
- Shin YW, Lee ST, Shin JW, et al. VGKC-complex/LGI1-antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol 2013; 265:75–81.
- Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013; 12:157–165.
- Brierley JB, Corsellis JAN, Hierons R, Nevin S. Subacute encephalitis of later adult life mainly affecting the limbic areas. Brain 1960; 83:357–368.
- Corsellis JA, Goldberg GJ, Norton AR. “Limbic encephalitis” and its association with carcinoma. Brain 1968; 91:481–496.
- Greenlee JE, Brashear HR. Antibodies to cerebellar Purkinje cells in patients with paraneoplastic cerebellar degeneration and ovarian carcinoma. Ann Neurol 1983; 14:609–613.
- Rosenfeld MR, Dalmau JO. Paraneoplastic disorders of the CNS and autoimmune synaptic encephalitis. Continuum (Minneap Minn) 2012; 18:366–383.
- Irani SR, Gelfand JM, Al-Diwani A, Vincent A. Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Ann Neurol 2014; 76:168–184.
- Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007; 61:25–36.
- Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 2010; 133:2734–2748.
- Lancaster E, Lai M, Peng X, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010; 9:67–76.
- Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009; 65:424–434.
- Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry 2012; 83:638–645.
- Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain 2005; 128:1764–1777.
- Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004; 127:701–712.
- Jammoul A, Lederman RJ, Tavee J, Li Y. Presence of voltage-gated potassium channel complex antibody in a case of genetic prion disease. BMJ Case Rep 2014; pii:bcr2013201622.
- Angus-Leppan H, Rudge P, Mead S, Collinge J, Vincent A. Autoantibodies in sporadic Creutzfeldt-Jakob disease. JAMA Neurol 2013; 70:919–922.
- Fujita K, Yuasa T, Watanabe O, et al. Voltage-gated potassium channel complex antibodies in Creutzfeldt-Jakob disease. J Neurol 2012; 259:2249–2250.
- Fujita K, Yuasa T, Takahashi Y, et al. Antibodies to N-methyl-D-aspartate glutamate receptors in Creutzfeldt–Jakob disease patients. J Neuroimmunol 2012; 251:90–93.
- Mackay G, Ahmad K, Stone J, et al. NMDA receptor autoantibodies in sporadic Creutzfeldt-Jakob disease. J Neurol 2012; 259:1979–1981.
- Leypoldt F, Titulaer MJ, Aguilar E, et al. Herpes simplex virus–1 encephalitis can trigger anti-NMDA receptor encephalitis: case report. Neurology 2013; 81:1637–1639.
- Desena A, Graves D, Warnack W, Greenberg BM. Herpes simplex encephalitis as a potential cause of anti-N-methyl-D-aspartate receptor antibody encephalitis: report of 2 cases. JAMA Neurol 2014; 71:344–346.
- Armangue T, Leypoldt F, Málaga I, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol 2014; 75:317–323.
- Blaes F. Paraneoplastic brain stem encephalitis. Curr Treat Options Neurol 2013; 15:201–209.
- Wildemann B, Jarius S. The expanding range of autoimmune disorders of the nervous system. Lancet Neurol 2013; 12:22–24.
- Kim W, Kim SH, Lee SH, Li XF, Kim HJ. Brain abnormalities as an initial manifestation of neuromyelitis optica spectrum disorder. Mult Scler 2011; 17:1107–1112.
- Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry 2013; 84:576–583.
- Jubelt B, Mihai C, Li MT, Veerapaneni P. Rhombencephalitis/brainstem encephalitis. Curr Neurol Neurosci Rep 2011; 11:543–552.
- Sillevis Smitt P, Kinoshita A, De Leeuw B, et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med 2000; 342:21–27.
- Ishida K, Mitoma H, Son SY, et al. Selective suppression of cerebellar GABAergic transmission by an autoantibody to glutamic acid decarboxylase. Ann Neurol 1999; 46:263–267.
- Hart IK, Waters C, Vincent A, et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol 1997; 41:238–246.
- Barber P, Anderson NE, Vincent A. Morvan’s syndrome associated with voltage-gated K+ channel antibodies. Neurology 2000; 54:771–772.
- Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001; 50:73–78.
- Majoie HJ, de Baets M, Renier W, Lang B, Vincent A. Antibodies to voltage-gated potassium and calcium channels in epilepsy. Epilepsy Res 2006; 71:135–141.
- Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology 2008; 70:1883–1890.
- Knowles CH, Lang B, Clover L, et al. A role for autoantibodies in some cases of acquired non-paraneoplastic gut dysmotility. Scand J Gastroenterol 2002; 37:166–170.
- Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011; 69:892–900.
- Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 2013: 136:3151–3162.
- Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol 2011; 10:759–772.
- Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011; 10:63–74.
- Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010; 133:1655–1667.
- Flanagan EP, McKeon A, Lennon VA, et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc 2010; 85:881–897.
- Younes-Mhenni S, Janier MF, Cinotti L, et al. FDG-PET improves tumour detection in patients with paraneoplastic neurological syndromes. Brain 2004; 127:2331–2338.
- Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011; 77:179–189.
- Shin YW, Lee ST, Shin JW, et al. VGKC-complex/LGI1-antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol 2013; 265:75–81.
- Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013; 12:157–165.
KEY POINTS
- Autoantibody-mediated encephalitis accounts for a portion of cases of unexplained status epilepticus, encephalitis, and acute-onset psychiatric symptoms.
- Magnetic resonance imaging and cerebrospinal fluid analysis may be normal early in the disease course.
- Patients can express more than one autoantibody and present with more than one neuronal syndrome.
- Syndromes in which antibodies attack antigens on the surface of neurons are more likely to respond to immunotherapy than those involving intracellular antigens.
- Anti-N-methyl-d-aspartate receptor encephalitis typically presents with psychosis, seizures, and movement disorders in young women and is often associated with an ovarian teratoma.
- Limbic encephalitis, mediated by antibody to the voltage-gated potassium channel complex, is typically nonneoplastic and responds well to immunotherapy.