User login
A breakthrough in understanding the pathogenesis of thrombotic thrombocytopenic purpura (TTP) came with the discovery of ADAMTS13 (an abbreviation for “a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13”), a plasma protein that cleaves von Willebrand factor, which interacts with platelets to promote blood clotting. If ADAMTS13 is lacking, unusually large multimers of von Willebrand factor can accumulate and trigger intravascular platelet aggregation and microthrombosis, causing the signs and symptoms of TTP.1–3
This knowledge has practical applications: we can now measure ADAMTS13 activity, ADAMTS13 inhibitor, and antibodies against ADAMTS13 to help us diagnose TTP and distinguish it from other forms of thrombotic microangiopathy, such as hemolytic-uremic syndrome, that have similar symptoms but require different treatment.
Using case studies, this article describes typical presentations of acute and relapsing TTP; the role of laboratory testing, including the ADAMTS13 assay; how to distinguish TTP from other conditions that present similarly; and how to manage this condition.
A HIGH RISK OF DEATH WITHOUT PLASMA EXCHANGE
TTP is characterized by disseminated microthrombi composed of agglutinated platelets and von Willebrand factor in small vessels. Tissue damage by microthrombi can cause thrombocytopenia (platelet deficiency), microangiopathic hemolytic anemia (loss of red blood cells caused by destructive conditions in small vessels), and multiorgan failure.1
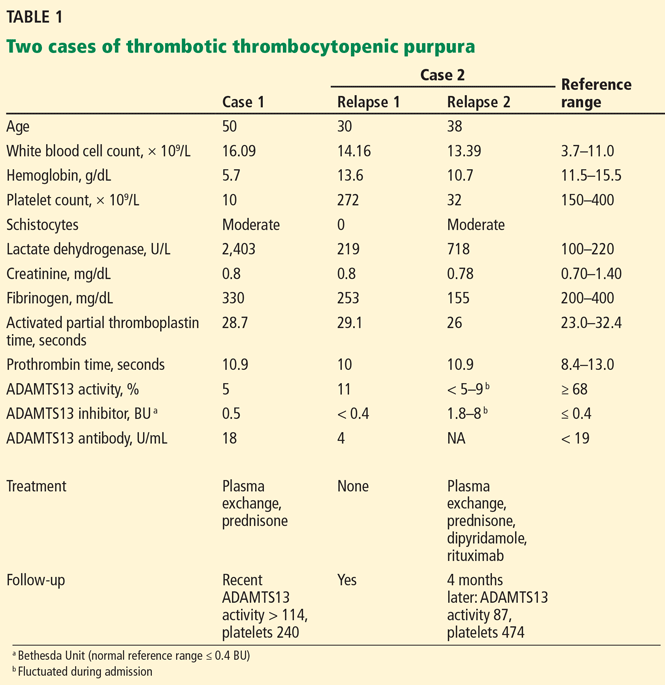
Untreated TTP has a mortality rate of about 90%.1 As shown in Case 1, Case 2, and Table 1, rapid diagnosis and prompt initiation of daily therapeutic plasma exchange can improve this grave outlook.4


ADAMTS13 DEFICIENCY CAN BE ACQUIRED OR CONGENITAL
Two major forms of TTP with ADAMTS13 deficiency and microvascular thrombosis are recognized:
Acquired TTP, the more common form, peaks in incidence between ages 30 and 50.2,5 It more often affects women, particularly during and after pregnancy (its estimated prevalence is 1 in 25,000 pregnancies), and African Americans.6 Acquired TTP may be:
- Primary (idiopathic or autoantibody-mediated), associated with severely decreased ADAMTS13 and the presence of ultra-large von Willebrand factor multimers, or
- Secondary (23%–67% of cases), arising from a variety of conditions, including autoimmune disorders (eg, systemic lupus erythematosus, rheumatoid arthritis), solid organ or hematopoietic cell transplant, malignancy, drugs, and pregnancy (Table 2).1,5–8 Secondary TTP has a worse prognosis than idiopathic TTP.5,9

Congenital TTP (Upshaw-Shulman syndrome) is a rare autosomal-recessive disease caused by compound heterozygous or homozygous mutations of the ADAMTS13 gene, producing nonfunctional ADAMTS13 protein. Patients have severely deficient ADAMTS13 activity but usually do not develop autoantibodies. There is a high risk of chronic, relapsing episodes; identified triggers include pregnancy and heavy alcohol intake.2,10 About half of patients with congenital TTP have an early onset, usually presenting with acute TTP between birth and age 5, and about half have a late onset, usually remaining without symptoms until age 20 to 40.
THE CLINICAL PICTURE OF TTP IS NOT ALWAYS CLASSIC
TTP is primarily diagnosed clinically, but diagnosis is often difficult because of various nonspecific symptoms. Typical TTP presents with the “classic pentad”:
- Severe thrombocytopenia (70%–100% of patients)
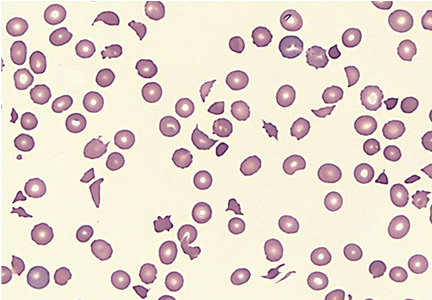
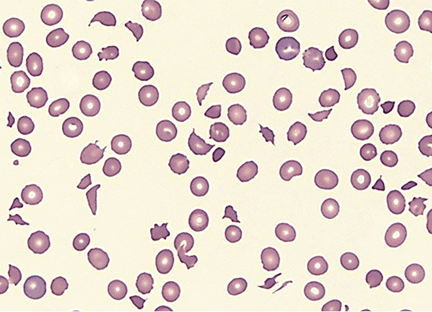
- Microangiopathic hemolytic anemia with multiple schistocytes (70%–100%) (Figure 1)
- Neurologic involvement (50%–90%)
- Renal abnormalities (about 50%)
- Fever (25%).
However, the entire picture often does not emerge in a single patient.2,6 Waiting for the entire pentad to develop before diagnosing TTP can have grave clinical consequences,1,2,5 and the presence of thrombocytopenia and unexplained microangiopathic hemolytic anemia are considered clinically sufficient to suspect TTP.5
Neurologic symptoms usually fluctuate. They can include mild abnormalities such as weakness, dizziness, headache, blurred vision, ataxia, and transient mental status changes, as well as severe abnormalities including stroke, seizure, and coma.2,6
Most patients have normal findings on computed tomography and magnetic resonance imaging at the onset of neurologic symptoms or with a history of TTP. Some patients (8%–39%) show reversible acute brain lesions, including ischemic changes.11–13
Other signs and symptoms may result from multiorgan failure due to microthrombosis; ischemia in retinal, coronary, and abdominal circulations; and unconjugated hyperbilirubinemia.2
Atypical presentations. About 18% of patients have cardiac involvement from microvascular occlusion, with arrhythmia, angina, or congestive heart failure. Abdominal pain and pancreatitis occur in 5% to 13%, and visual disturbances in 8% to 10%.
Patients with an atypical presentation may not have laboratory evidence of microangiopathic hemolytic anemia, but an ADAMTS13 assay will show severely decreased activity. Therapeutic plasma exchange can improve atypical symptoms.2,3,10,14,15
ADAMTS13 ASSAY IS KEY TO DIAGNOSIS
Laboratory evidence typically includes hemolytic anemia (reticulocytosis, schistocytes, elevated indirect bilirubin, reduced haptoglobin, elevated lactate dehydrogenase) and thrombocytopenia.3 There are no significant abnormalities in prothrombin time, international normalized ratio, activated partial thromboplastin time, fibrinogen, or D-dimer level.
Measuring the levels of ADAMTS13 activity, ADAMTS13 inhibitor, and ADAMTS13 antibody is becoming standard to confirm the diagnosis of TTP, to determine if it is congenital or acquired, and to distinguish it from thrombocytopenic conditions such as hemolytic-uremic syndrome, idiopathic thrombocytopenic purpura, and heparin-induced thrombocytopenia.4,5 A newer ADAMTS13 assay based on fluorescence energy transfer (FRET) technology with a synthetic amino acid-von Willebrand factor peptide substrate has a faster turnaround time and less test variability.6,16,17 This FRET assay can give the result of ADAMTS13 activity within 2 hours. In comparison, the assay based on multimeric von Willebrand factor takes 2 to 3 days, and mass spectrometry to measure the cleavage products of a synthetic von Willebrand factor molecule takes about 4 hours.3,10,16
About two-thirds of patients with the clinical diagnosis of idiopathic TTP have ADAMTS13 activity levels lower than 10%.5,14,18 In the appropriate clinical setting, this threshold level is highly sensitive (89%–100%) and specific (99%–100%) in differentiating TTP from other thrombotic angiopathies.2,3,18
Note: The ADAMTS13 assay was needed for early correct diagnosis in Case 1 and Case 2.
Inhibitors provide more clues
Autoantibodies can be classified according to whether they inhibit ADAMTS13 activity.
Neutralizing inhibitors. Most cases of acquired, idiopathic TTP with severe ADAMTS13 deficiency are related to circulating autoantibodies that neutralize ADAMTS13 activity. This ADAMTS13 inhibitor level is obtained by measuring residual ADAMTS13 activity after mixing equal amounts of patient plasma with normal pooled plasma. ADAMTS13 inhibitor is detectable in 44% to 93% of patients with severely deficient ADAMTS13 activity.3,6,19
Nonneutralizing inhibitors. From 10% to 15% of patients with TTP with severe ADAMTS13 deficiency lack ADAMTS13 autoantibodies measured by enzyme immunoassay but have nonneutralizing immunoglobulin G (IgG) or IgM autoantibodies. In such cases, ADAMTS13 deficiency may be related to increased antibody-mediated clearance or other unknown mechanisms.
Neutralizing inhibitors and nonneutralizing inhibitors may be present simultaneously in some patients.3,10,19,20
Blood factors affect ADAMTS13 activity
Specimen factors can affect ADAMTS13 activity and antibody levels.
Hemoglobin is a potent inhibitor of ADAMTS13, so an elevated plasma level of free hemoglobin (> 2 g/dL) can reduce ADAMTS13 activity, as can hyperbilirubinemia (> 15 mg/dL).
High levels of endogenous von Willebrand factor, lipids, thrombin, or other proteases that may cleave ADAMTS13 can also reduce ADAMTS13 activity.3 Conversely, recent plasma exchange or transfusion can mask the diagnosis of TTP because of false normalization of ADAMTS13 activity. In addition, ADAMTS13 autoantibody can be detected in other immune-mediated disorders (eg, systemic lupus erythematosus, antiphospholipid syndrome), and hypergammaglobulinemia, as well as in 10% to 15% of healthy individuals.19
CONSIDER OTHER CONDITIONS

Before diagnosing TTP, other conditions causing thrombocytopenia and hemolytic anemia should be excluded by taking a careful clinical, laboratory, and medication history (Table 2). Of these conditions, the most challenging to differentiate from TTP—and often indistinguishable from it at presentation—is hemolytic-uremic syndrome (Table 3).
Hemolytic-uremic syndrome
Hemolytic-uremic syndrome presents with a triad of thrombocytopenia, acute renal failure, and microangiopathic hemolytic anemia, with increased lactate dehydrogenase levels. Renal dysfunction from ischemia or tissue injury by microvascular thrombi predominates. Hemolytic-uremic syndrome most often occurs in children and is often related to hemorrhagic enterocolitis caused by infection with Escherichia coli O157:H7 or Shigella species (90%–95% of cases).1,2,5
From 5% to 10% of cases of hemolytic- uremic syndrome are atypical. These cases are not associated with diarrhea, and many are caused by genetic mutations that result in chronic excessive complement activation. Implicated genes regulate complement regulator factor H (20%–30% of cases) or CD46 (10%) and other cofactors, or autoantibodies against factor H (10%), which affect the alternate complement pathway.6,21–23
Initial therapeutic plasma exchange is commonly undertaken for atypical hemolytic- uremic syndrome, particularly for patients at risk of rapid progression to end-stage renal failure. But despite such treatment, about 60% of these patients die or develop permanent renal damage within 1 year.2,3,24
Eculizumab, a monoclonal antibody against complement component C5, has been approved by the US Food and Drug Administration for atypical hemolytic-uremic syndrome and may improve quality of life.25–27
PLASMA EXCHANGE IS THE MAINSTAY OF THERAPY
In 2012, the British Society for Haematology published revised guidelines for managing TTP and other thrombotic microangiopathies.28
Acquired idiopathic TTP with reduced ADAMTS13 activity requires immediate therapeutic plasma exchange. Daily plasma exchange combines plasmapheresis to remove circulating ultralarge von Willebrand factor-platelet strings and autoantibodies against ADAMTS13, and infusion of fresh-frozen plasma to replace ADAMTS13.18 This procedure is the mainstay of therapy and brings 70% to 90% of patients with idiopathic TTP to remission.1,2,5,6 However, the optimal duration of daily plasma exchange and the number of procedures required is highly variable according to clinical condition. Therapeutic plasma exchange can also cause plasma-related adverse reactions.9,28 Congenital TTP requires plasma infusion or exchange depending on the patient’s severity of ADAMTS13 deficiency.
Corticosteroids are used in combination with daily therapeutic plasma exchange, although evidence from controlled trials of their efficacy in this setting is lacking. Patients with severely decreased ADAMTS13 activity or low titers of ADAMTS13 autoantibodies tend to respond to the therapy.5,8,29
An ADAMTS13 assay with a short turn-around time can help guide the decision to initiate therapeutic plasma exchange. However, if there is a strong clinical suspicion of TTP, plasma exchange should be initiated immediately without waiting for test results.5,30 Monitoring ADAMTS13 activity or inhibitor during initial plasma exchange therapy has had conflicting results in several studies and is generally not recommended for patients with acquired TTP.8,30,31
RELAPSE IS COMMON
About 20% to 50% of patients with idiopathic TTP experience a relapse (Case 2). Most relapses occur within the first 2 years after the initial episode, with an estimated risk of 43% for relapse at 7.5 years.5,9
Factors that predict a higher risk of relapse include persistently severely decreased ADAMTS13 activity, positive inhibitor, and high titers of autoantibodies to ADAMTS13 during symptomatic TTP. During clinical remission, persistence of autoantibodies also indicates increased risk.1,3,5,6,9
Patients who have a relapse and whose disease is refractory to therapeutic plasma exchange (10%–20% of cases) have been treated with corticosteroids, splenectomy, or immunosuppressive agents (cyclosporine, azathioprine, or cyclophosphamide) with varying rates of success. Rituximab (monoclonal anti-CD20) has recently been used as second-line therapy in refractory or relapsing immune-mediated TTP or idiopathic TTP with neurologic or cardiac symptoms associated with a poor prognosis. Therapy including rituximab results in improved response and progression-free survival.32 Other potential therapies, including recombinant active ADAMTS13, are under investigation.9,23,28,30,33,34
- Sadler JE, Moake JL, Miyata T, George JN. Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2004; 1:407–423.
- Shenkman B, Einav Y. Thrombotic thrombocytopenic purpura and other thrombotic microangiopathic hemolytic anemias: diagnosis and classification. Autoimmun Rev 2014; 13:584–586.
- Shah N, Sarode R. Thrombotic thrombocytopenic purpura-what is new? J Clin Apher 2013; 28:30–35.
- Imanirad I, Rajasekhar A, Zumberg M. A case series of atypical presentations of thrombotic thrombocytopenic purpura. J Clin Apher 2012; 27:221–226.
- George JN, Al-Nouri ZL. Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes. Hematology Am Soc Hematol Educ Program 2012; 1:604–609.
- Shah N, Rutherford C, Matevosyan K, Shen YM, Sarode R. Role of ADAMTS13 in the management of thrombotic microangiopathies including thrombotic thrombocytopenic purpura (TTP). Br J Haematol 2013; 163:514–519.
- Cataland SR, Yang S, Wu HM. The use of ADAMTS13 activity, platelet count, and serum creatinine to differentiate acquired thrombotic thrombocytopenic purpura from other thrombotic microangiopathies. Br J Haematol 2012; 157:501–503.
- Mannucci PM, Peyvandi F. TTP and ADAMTS13: when Is testing appropriate? Hematology Am Soc Hematol Educ Program 2007; 1:121–126.
- Chaturved S, Carcioppolo D, Zhang L, McCar KR. Management and outcomes of patients with TTP: analysis of 100 cases at a single institution. Am J Hematol 2013; 88:560–565.
- Peyvandi F, Palla R, Lotta LA, Mackie I, Scully MA, Machin SJ. ADAMTS-13 assays in thrombotic thrombocytopenic purpura. J Thromb Haemost 2010; 8:631–640.
- Cataland SR, Scully MA, Paskavitz J, et al. Evidence of persistent neurologic injury following thrombotic thrombocytopenic purpura. Am J Hematol 2011; 86:87–89.
- Meloni G, Proia A, Antonini G, et al. Thrombotic thrombocytopenic purpura: prospective neurologic, neuroimaging and neurophysiologic evaluation. Haematologica 2001; 86:1194–1199.
- Kwaan HC, Boggio LN. The clinical spectrum of thrombotic thrombocytopenic purpura. Semin Thromb Hemost 2005; 31:673–680.
- Sarode R. Atypical presentations of thrombotic thrombocytopenic purpura: a review. J Clin Apher 2009; 24:47–52.
- Volcy J, Nzerue CM, Oderinde A, Hewan-Iowe K. Cocaine-induced acute renal failure, hemolysis, and thrombocytopenia mimicking thrombotic thrombocytopenic purpura. Am J Kidney Dis 2000; 35:E3.
- Kremer Hovinga JA, Mottini M, Lammle B. Measurement of ADAMTS-13 activity in plasma by the FRETS-VWF73 assay: comparison with other assay methods. J Thromb Haemost 2006; 4:1146–1148.
- Groot E, Hulstein JJ, Rison CN, de Groot PG, Fijnheer R. FRETS-VWF73: a rapid and predictive tool for thrombotic thrombocytopenic purpura. J Thromb Haemost 2006; 4:698–699.
- Barrows BD, Teruya J. Use of the ADAMTS13 activity assay improved the accuracy and efficiency of the diagnosis and treatment of suspected acquired thrombotic thrombocytopenic purpura. Arch Pathol Lab Med 2014; 138:546–549.
- Rieger M, Mannucci PM, Kremer Hovinga JA, et al. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood 2005; 106:1262–1267.
- Rogers HJ, Kottke-Marchant K. ADAMTS13 evaluation for thrombotic thrombocytopenic purpura. Pathology Innovations, Pathology and Laboratory Medicine Institute. Cleveland Clinic, Fall 2014:6–9.
- Józsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008; 111:1512–1514.
- Diamante Chiodini B, Davin JC, Corazza F, et al. Eculizumab in anti-factor H antibodies associated with atypical hemolytic uremic syndrome. Pediatrics 2014; 133:e1764–e1768.
- Taylor CM, Machin S, Wigmore SJ, Goodship TH; working party from the Renal Association, the British Committee for Standards in Haematology and the British Transplantation Society. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol 2009; 148:37–47.
- Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 2010; 36:673–681.
- Tsai HM, Kuo E. Eculizumab therapy leads to rapid resolution of thrombocytopenia in atypical hemolytic uremic syndrome. Adv Hematol 2014; 295323:1–7.
- Lapeyraque AL, Frémeaux-Bacchi V, Robitaille P. Efficacy of eculizumab in a patient with factor-H-associated atypical hemolytic uremic syndrome. Pediatr Nephrol 2011; 26:621–624.
- Baskin E, Gulleroglu K, Kantar A, Bayrakci U, Ozkaya O. Success of eculizumab in the treatment of atypical hemolytic uremic syndrome. Pediatr Nephrol 2015; 30:783–789.
- Scully M, Hunt BJ, Benjamin S, et al; British Committee for Standards in Haematology. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 2012; 158:323–325.
- Abassi E, Yawn D, Leveque E, Nolasco L, Lopez J, Moake J. Correlation of ADAMTS-13 activity with response to plasma exchange in patients diagnosed with thrombotic thrombocytopenic purpura (Abstract #3921). Blood 2004; 104:242a.
- Blombery P, Scully M. Management of thrombocytic thrombocytopenic purpura: current perspectives. J Blood Med 2014; 5:15–23.
- Wu N, Liu J, Yang S, et al. Diagnostic and prognostic values of ADAMTS13 activity measured during daily plasma exchange therapy in patients with acquired thrombotic thrombocytopenic purpura. Transfusion 2015; 55:18–24.
- Cuker A. Adjuvant rituximab to prevent TTP relapse. Blood 2016; 127:2952–2953.
- Chapman K, Yuen S. Therapy for thrombotic thrombocytopenic purpura: past, present and future. Semin Thromb Hemost 2014; 40:34–40.
- Heidel F, Lipka DB, von Auer C, Huber C, Schrarrer I, Hess G. Addition of rituximab to standard therapy improves response rate and progression-free survival in relapsed or refractory thrombotic thrombocytopenic purpura and autoimmune haemolytic anaemia. Thromb Haemost 2007; 97:228–233.
A breakthrough in understanding the pathogenesis of thrombotic thrombocytopenic purpura (TTP) came with the discovery of ADAMTS13 (an abbreviation for “a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13”), a plasma protein that cleaves von Willebrand factor, which interacts with platelets to promote blood clotting. If ADAMTS13 is lacking, unusually large multimers of von Willebrand factor can accumulate and trigger intravascular platelet aggregation and microthrombosis, causing the signs and symptoms of TTP.1–3
This knowledge has practical applications: we can now measure ADAMTS13 activity, ADAMTS13 inhibitor, and antibodies against ADAMTS13 to help us diagnose TTP and distinguish it from other forms of thrombotic microangiopathy, such as hemolytic-uremic syndrome, that have similar symptoms but require different treatment.
Using case studies, this article describes typical presentations of acute and relapsing TTP; the role of laboratory testing, including the ADAMTS13 assay; how to distinguish TTP from other conditions that present similarly; and how to manage this condition.
A HIGH RISK OF DEATH WITHOUT PLASMA EXCHANGE
TTP is characterized by disseminated microthrombi composed of agglutinated platelets and von Willebrand factor in small vessels. Tissue damage by microthrombi can cause thrombocytopenia (platelet deficiency), microangiopathic hemolytic anemia (loss of red blood cells caused by destructive conditions in small vessels), and multiorgan failure.1
Untreated TTP has a mortality rate of about 90%.1 As shown in Case 1, Case 2, and Table 1, rapid diagnosis and prompt initiation of daily therapeutic plasma exchange can improve this grave outlook.4
ADAMTS13 DEFICIENCY CAN BE ACQUIRED OR CONGENITAL
Two major forms of TTP with ADAMTS13 deficiency and microvascular thrombosis are recognized:
Acquired TTP, the more common form, peaks in incidence between ages 30 and 50.2,5 It more often affects women, particularly during and after pregnancy (its estimated prevalence is 1 in 25,000 pregnancies), and African Americans.6 Acquired TTP may be:
- Primary (idiopathic or autoantibody-mediated), associated with severely decreased ADAMTS13 and the presence of ultra-large von Willebrand factor multimers, or
- Secondary (23%–67% of cases), arising from a variety of conditions, including autoimmune disorders (eg, systemic lupus erythematosus, rheumatoid arthritis), solid organ or hematopoietic cell transplant, malignancy, drugs, and pregnancy (Table 2).1,5–8 Secondary TTP has a worse prognosis than idiopathic TTP.5,9
Congenital TTP (Upshaw-Shulman syndrome) is a rare autosomal-recessive disease caused by compound heterozygous or homozygous mutations of the ADAMTS13 gene, producing nonfunctional ADAMTS13 protein. Patients have severely deficient ADAMTS13 activity but usually do not develop autoantibodies. There is a high risk of chronic, relapsing episodes; identified triggers include pregnancy and heavy alcohol intake.2,10 About half of patients with congenital TTP have an early onset, usually presenting with acute TTP between birth and age 5, and about half have a late onset, usually remaining without symptoms until age 20 to 40.
THE CLINICAL PICTURE OF TTP IS NOT ALWAYS CLASSIC
TTP is primarily diagnosed clinically, but diagnosis is often difficult because of various nonspecific symptoms. Typical TTP presents with the “classic pentad”:
- Severe thrombocytopenia (70%–100% of patients)
- Microangiopathic hemolytic anemia with multiple schistocytes (70%–100%) (Figure 1)
- Neurologic involvement (50%–90%)
- Renal abnormalities (about 50%)
- Fever (25%).
However, the entire picture often does not emerge in a single patient.2,6 Waiting for the entire pentad to develop before diagnosing TTP can have grave clinical consequences,1,2,5 and the presence of thrombocytopenia and unexplained microangiopathic hemolytic anemia are considered clinically sufficient to suspect TTP.5
Neurologic symptoms usually fluctuate. They can include mild abnormalities such as weakness, dizziness, headache, blurred vision, ataxia, and transient mental status changes, as well as severe abnormalities including stroke, seizure, and coma.2,6
Most patients have normal findings on computed tomography and magnetic resonance imaging at the onset of neurologic symptoms or with a history of TTP. Some patients (8%–39%) show reversible acute brain lesions, including ischemic changes.11–13
Other signs and symptoms may result from multiorgan failure due to microthrombosis; ischemia in retinal, coronary, and abdominal circulations; and unconjugated hyperbilirubinemia.2
Atypical presentations. About 18% of patients have cardiac involvement from microvascular occlusion, with arrhythmia, angina, or congestive heart failure. Abdominal pain and pancreatitis occur in 5% to 13%, and visual disturbances in 8% to 10%.
Patients with an atypical presentation may not have laboratory evidence of microangiopathic hemolytic anemia, but an ADAMTS13 assay will show severely decreased activity. Therapeutic plasma exchange can improve atypical symptoms.2,3,10,14,15
ADAMTS13 ASSAY IS KEY TO DIAGNOSIS
Laboratory evidence typically includes hemolytic anemia (reticulocytosis, schistocytes, elevated indirect bilirubin, reduced haptoglobin, elevated lactate dehydrogenase) and thrombocytopenia.3 There are no significant abnormalities in prothrombin time, international normalized ratio, activated partial thromboplastin time, fibrinogen, or D-dimer level.
Measuring the levels of ADAMTS13 activity, ADAMTS13 inhibitor, and ADAMTS13 antibody is becoming standard to confirm the diagnosis of TTP, to determine if it is congenital or acquired, and to distinguish it from thrombocytopenic conditions such as hemolytic-uremic syndrome, idiopathic thrombocytopenic purpura, and heparin-induced thrombocytopenia.4,5 A newer ADAMTS13 assay based on fluorescence energy transfer (FRET) technology with a synthetic amino acid-von Willebrand factor peptide substrate has a faster turnaround time and less test variability.6,16,17 This FRET assay can give the result of ADAMTS13 activity within 2 hours. In comparison, the assay based on multimeric von Willebrand factor takes 2 to 3 days, and mass spectrometry to measure the cleavage products of a synthetic von Willebrand factor molecule takes about 4 hours.3,10,16
About two-thirds of patients with the clinical diagnosis of idiopathic TTP have ADAMTS13 activity levels lower than 10%.5,14,18 In the appropriate clinical setting, this threshold level is highly sensitive (89%–100%) and specific (99%–100%) in differentiating TTP from other thrombotic angiopathies.2,3,18
Note: The ADAMTS13 assay was needed for early correct diagnosis in Case 1 and Case 2.
Inhibitors provide more clues
Autoantibodies can be classified according to whether they inhibit ADAMTS13 activity.
Neutralizing inhibitors. Most cases of acquired, idiopathic TTP with severe ADAMTS13 deficiency are related to circulating autoantibodies that neutralize ADAMTS13 activity. This ADAMTS13 inhibitor level is obtained by measuring residual ADAMTS13 activity after mixing equal amounts of patient plasma with normal pooled plasma. ADAMTS13 inhibitor is detectable in 44% to 93% of patients with severely deficient ADAMTS13 activity.3,6,19
Nonneutralizing inhibitors. From 10% to 15% of patients with TTP with severe ADAMTS13 deficiency lack ADAMTS13 autoantibodies measured by enzyme immunoassay but have nonneutralizing immunoglobulin G (IgG) or IgM autoantibodies. In such cases, ADAMTS13 deficiency may be related to increased antibody-mediated clearance or other unknown mechanisms.
Neutralizing inhibitors and nonneutralizing inhibitors may be present simultaneously in some patients.3,10,19,20
Blood factors affect ADAMTS13 activity
Specimen factors can affect ADAMTS13 activity and antibody levels.
Hemoglobin is a potent inhibitor of ADAMTS13, so an elevated plasma level of free hemoglobin (> 2 g/dL) can reduce ADAMTS13 activity, as can hyperbilirubinemia (> 15 mg/dL).
High levels of endogenous von Willebrand factor, lipids, thrombin, or other proteases that may cleave ADAMTS13 can also reduce ADAMTS13 activity.3 Conversely, recent plasma exchange or transfusion can mask the diagnosis of TTP because of false normalization of ADAMTS13 activity. In addition, ADAMTS13 autoantibody can be detected in other immune-mediated disorders (eg, systemic lupus erythematosus, antiphospholipid syndrome), and hypergammaglobulinemia, as well as in 10% to 15% of healthy individuals.19
CONSIDER OTHER CONDITIONS
Before diagnosing TTP, other conditions causing thrombocytopenia and hemolytic anemia should be excluded by taking a careful clinical, laboratory, and medication history (Table 2). Of these conditions, the most challenging to differentiate from TTP—and often indistinguishable from it at presentation—is hemolytic-uremic syndrome (Table 3).
Hemolytic-uremic syndrome
Hemolytic-uremic syndrome presents with a triad of thrombocytopenia, acute renal failure, and microangiopathic hemolytic anemia, with increased lactate dehydrogenase levels. Renal dysfunction from ischemia or tissue injury by microvascular thrombi predominates. Hemolytic-uremic syndrome most often occurs in children and is often related to hemorrhagic enterocolitis caused by infection with Escherichia coli O157:H7 or Shigella species (90%–95% of cases).1,2,5
From 5% to 10% of cases of hemolytic- uremic syndrome are atypical. These cases are not associated with diarrhea, and many are caused by genetic mutations that result in chronic excessive complement activation. Implicated genes regulate complement regulator factor H (20%–30% of cases) or CD46 (10%) and other cofactors, or autoantibodies against factor H (10%), which affect the alternate complement pathway.6,21–23
Initial therapeutic plasma exchange is commonly undertaken for atypical hemolytic- uremic syndrome, particularly for patients at risk of rapid progression to end-stage renal failure. But despite such treatment, about 60% of these patients die or develop permanent renal damage within 1 year.2,3,24
Eculizumab, a monoclonal antibody against complement component C5, has been approved by the US Food and Drug Administration for atypical hemolytic-uremic syndrome and may improve quality of life.25–27
PLASMA EXCHANGE IS THE MAINSTAY OF THERAPY
In 2012, the British Society for Haematology published revised guidelines for managing TTP and other thrombotic microangiopathies.28
Acquired idiopathic TTP with reduced ADAMTS13 activity requires immediate therapeutic plasma exchange. Daily plasma exchange combines plasmapheresis to remove circulating ultralarge von Willebrand factor-platelet strings and autoantibodies against ADAMTS13, and infusion of fresh-frozen plasma to replace ADAMTS13.18 This procedure is the mainstay of therapy and brings 70% to 90% of patients with idiopathic TTP to remission.1,2,5,6 However, the optimal duration of daily plasma exchange and the number of procedures required is highly variable according to clinical condition. Therapeutic plasma exchange can also cause plasma-related adverse reactions.9,28 Congenital TTP requires plasma infusion or exchange depending on the patient’s severity of ADAMTS13 deficiency.
Corticosteroids are used in combination with daily therapeutic plasma exchange, although evidence from controlled trials of their efficacy in this setting is lacking. Patients with severely decreased ADAMTS13 activity or low titers of ADAMTS13 autoantibodies tend to respond to the therapy.5,8,29
An ADAMTS13 assay with a short turn-around time can help guide the decision to initiate therapeutic plasma exchange. However, if there is a strong clinical suspicion of TTP, plasma exchange should be initiated immediately without waiting for test results.5,30 Monitoring ADAMTS13 activity or inhibitor during initial plasma exchange therapy has had conflicting results in several studies and is generally not recommended for patients with acquired TTP.8,30,31
RELAPSE IS COMMON
About 20% to 50% of patients with idiopathic TTP experience a relapse (Case 2). Most relapses occur within the first 2 years after the initial episode, with an estimated risk of 43% for relapse at 7.5 years.5,9
Factors that predict a higher risk of relapse include persistently severely decreased ADAMTS13 activity, positive inhibitor, and high titers of autoantibodies to ADAMTS13 during symptomatic TTP. During clinical remission, persistence of autoantibodies also indicates increased risk.1,3,5,6,9
Patients who have a relapse and whose disease is refractory to therapeutic plasma exchange (10%–20% of cases) have been treated with corticosteroids, splenectomy, or immunosuppressive agents (cyclosporine, azathioprine, or cyclophosphamide) with varying rates of success. Rituximab (monoclonal anti-CD20) has recently been used as second-line therapy in refractory or relapsing immune-mediated TTP or idiopathic TTP with neurologic or cardiac symptoms associated with a poor prognosis. Therapy including rituximab results in improved response and progression-free survival.32 Other potential therapies, including recombinant active ADAMTS13, are under investigation.9,23,28,30,33,34
A breakthrough in understanding the pathogenesis of thrombotic thrombocytopenic purpura (TTP) came with the discovery of ADAMTS13 (an abbreviation for “a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13”), a plasma protein that cleaves von Willebrand factor, which interacts with platelets to promote blood clotting. If ADAMTS13 is lacking, unusually large multimers of von Willebrand factor can accumulate and trigger intravascular platelet aggregation and microthrombosis, causing the signs and symptoms of TTP.1–3
This knowledge has practical applications: we can now measure ADAMTS13 activity, ADAMTS13 inhibitor, and antibodies against ADAMTS13 to help us diagnose TTP and distinguish it from other forms of thrombotic microangiopathy, such as hemolytic-uremic syndrome, that have similar symptoms but require different treatment.
Using case studies, this article describes typical presentations of acute and relapsing TTP; the role of laboratory testing, including the ADAMTS13 assay; how to distinguish TTP from other conditions that present similarly; and how to manage this condition.
A HIGH RISK OF DEATH WITHOUT PLASMA EXCHANGE
TTP is characterized by disseminated microthrombi composed of agglutinated platelets and von Willebrand factor in small vessels. Tissue damage by microthrombi can cause thrombocytopenia (platelet deficiency), microangiopathic hemolytic anemia (loss of red blood cells caused by destructive conditions in small vessels), and multiorgan failure.1
Untreated TTP has a mortality rate of about 90%.1 As shown in Case 1, Case 2, and Table 1, rapid diagnosis and prompt initiation of daily therapeutic plasma exchange can improve this grave outlook.4
ADAMTS13 DEFICIENCY CAN BE ACQUIRED OR CONGENITAL
Two major forms of TTP with ADAMTS13 deficiency and microvascular thrombosis are recognized:
Acquired TTP, the more common form, peaks in incidence between ages 30 and 50.2,5 It more often affects women, particularly during and after pregnancy (its estimated prevalence is 1 in 25,000 pregnancies), and African Americans.6 Acquired TTP may be:
- Primary (idiopathic or autoantibody-mediated), associated with severely decreased ADAMTS13 and the presence of ultra-large von Willebrand factor multimers, or
- Secondary (23%–67% of cases), arising from a variety of conditions, including autoimmune disorders (eg, systemic lupus erythematosus, rheumatoid arthritis), solid organ or hematopoietic cell transplant, malignancy, drugs, and pregnancy (Table 2).1,5–8 Secondary TTP has a worse prognosis than idiopathic TTP.5,9
Congenital TTP (Upshaw-Shulman syndrome) is a rare autosomal-recessive disease caused by compound heterozygous or homozygous mutations of the ADAMTS13 gene, producing nonfunctional ADAMTS13 protein. Patients have severely deficient ADAMTS13 activity but usually do not develop autoantibodies. There is a high risk of chronic, relapsing episodes; identified triggers include pregnancy and heavy alcohol intake.2,10 About half of patients with congenital TTP have an early onset, usually presenting with acute TTP between birth and age 5, and about half have a late onset, usually remaining without symptoms until age 20 to 40.
THE CLINICAL PICTURE OF TTP IS NOT ALWAYS CLASSIC
TTP is primarily diagnosed clinically, but diagnosis is often difficult because of various nonspecific symptoms. Typical TTP presents with the “classic pentad”:
- Severe thrombocytopenia (70%–100% of patients)
- Microangiopathic hemolytic anemia with multiple schistocytes (70%–100%) (Figure 1)
- Neurologic involvement (50%–90%)
- Renal abnormalities (about 50%)
- Fever (25%).
However, the entire picture often does not emerge in a single patient.2,6 Waiting for the entire pentad to develop before diagnosing TTP can have grave clinical consequences,1,2,5 and the presence of thrombocytopenia and unexplained microangiopathic hemolytic anemia are considered clinically sufficient to suspect TTP.5
Neurologic symptoms usually fluctuate. They can include mild abnormalities such as weakness, dizziness, headache, blurred vision, ataxia, and transient mental status changes, as well as severe abnormalities including stroke, seizure, and coma.2,6
Most patients have normal findings on computed tomography and magnetic resonance imaging at the onset of neurologic symptoms or with a history of TTP. Some patients (8%–39%) show reversible acute brain lesions, including ischemic changes.11–13
Other signs and symptoms may result from multiorgan failure due to microthrombosis; ischemia in retinal, coronary, and abdominal circulations; and unconjugated hyperbilirubinemia.2
Atypical presentations. About 18% of patients have cardiac involvement from microvascular occlusion, with arrhythmia, angina, or congestive heart failure. Abdominal pain and pancreatitis occur in 5% to 13%, and visual disturbances in 8% to 10%.
Patients with an atypical presentation may not have laboratory evidence of microangiopathic hemolytic anemia, but an ADAMTS13 assay will show severely decreased activity. Therapeutic plasma exchange can improve atypical symptoms.2,3,10,14,15
ADAMTS13 ASSAY IS KEY TO DIAGNOSIS
Laboratory evidence typically includes hemolytic anemia (reticulocytosis, schistocytes, elevated indirect bilirubin, reduced haptoglobin, elevated lactate dehydrogenase) and thrombocytopenia.3 There are no significant abnormalities in prothrombin time, international normalized ratio, activated partial thromboplastin time, fibrinogen, or D-dimer level.
Measuring the levels of ADAMTS13 activity, ADAMTS13 inhibitor, and ADAMTS13 antibody is becoming standard to confirm the diagnosis of TTP, to determine if it is congenital or acquired, and to distinguish it from thrombocytopenic conditions such as hemolytic-uremic syndrome, idiopathic thrombocytopenic purpura, and heparin-induced thrombocytopenia.4,5 A newer ADAMTS13 assay based on fluorescence energy transfer (FRET) technology with a synthetic amino acid-von Willebrand factor peptide substrate has a faster turnaround time and less test variability.6,16,17 This FRET assay can give the result of ADAMTS13 activity within 2 hours. In comparison, the assay based on multimeric von Willebrand factor takes 2 to 3 days, and mass spectrometry to measure the cleavage products of a synthetic von Willebrand factor molecule takes about 4 hours.3,10,16
About two-thirds of patients with the clinical diagnosis of idiopathic TTP have ADAMTS13 activity levels lower than 10%.5,14,18 In the appropriate clinical setting, this threshold level is highly sensitive (89%–100%) and specific (99%–100%) in differentiating TTP from other thrombotic angiopathies.2,3,18
Note: The ADAMTS13 assay was needed for early correct diagnosis in Case 1 and Case 2.
Inhibitors provide more clues
Autoantibodies can be classified according to whether they inhibit ADAMTS13 activity.
Neutralizing inhibitors. Most cases of acquired, idiopathic TTP with severe ADAMTS13 deficiency are related to circulating autoantibodies that neutralize ADAMTS13 activity. This ADAMTS13 inhibitor level is obtained by measuring residual ADAMTS13 activity after mixing equal amounts of patient plasma with normal pooled plasma. ADAMTS13 inhibitor is detectable in 44% to 93% of patients with severely deficient ADAMTS13 activity.3,6,19
Nonneutralizing inhibitors. From 10% to 15% of patients with TTP with severe ADAMTS13 deficiency lack ADAMTS13 autoantibodies measured by enzyme immunoassay but have nonneutralizing immunoglobulin G (IgG) or IgM autoantibodies. In such cases, ADAMTS13 deficiency may be related to increased antibody-mediated clearance or other unknown mechanisms.
Neutralizing inhibitors and nonneutralizing inhibitors may be present simultaneously in some patients.3,10,19,20
Blood factors affect ADAMTS13 activity
Specimen factors can affect ADAMTS13 activity and antibody levels.
Hemoglobin is a potent inhibitor of ADAMTS13, so an elevated plasma level of free hemoglobin (> 2 g/dL) can reduce ADAMTS13 activity, as can hyperbilirubinemia (> 15 mg/dL).
High levels of endogenous von Willebrand factor, lipids, thrombin, or other proteases that may cleave ADAMTS13 can also reduce ADAMTS13 activity.3 Conversely, recent plasma exchange or transfusion can mask the diagnosis of TTP because of false normalization of ADAMTS13 activity. In addition, ADAMTS13 autoantibody can be detected in other immune-mediated disorders (eg, systemic lupus erythematosus, antiphospholipid syndrome), and hypergammaglobulinemia, as well as in 10% to 15% of healthy individuals.19
CONSIDER OTHER CONDITIONS
Before diagnosing TTP, other conditions causing thrombocytopenia and hemolytic anemia should be excluded by taking a careful clinical, laboratory, and medication history (Table 2). Of these conditions, the most challenging to differentiate from TTP—and often indistinguishable from it at presentation—is hemolytic-uremic syndrome (Table 3).
Hemolytic-uremic syndrome
Hemolytic-uremic syndrome presents with a triad of thrombocytopenia, acute renal failure, and microangiopathic hemolytic anemia, with increased lactate dehydrogenase levels. Renal dysfunction from ischemia or tissue injury by microvascular thrombi predominates. Hemolytic-uremic syndrome most often occurs in children and is often related to hemorrhagic enterocolitis caused by infection with Escherichia coli O157:H7 or Shigella species (90%–95% of cases).1,2,5
From 5% to 10% of cases of hemolytic- uremic syndrome are atypical. These cases are not associated with diarrhea, and many are caused by genetic mutations that result in chronic excessive complement activation. Implicated genes regulate complement regulator factor H (20%–30% of cases) or CD46 (10%) and other cofactors, or autoantibodies against factor H (10%), which affect the alternate complement pathway.6,21–23
Initial therapeutic plasma exchange is commonly undertaken for atypical hemolytic- uremic syndrome, particularly for patients at risk of rapid progression to end-stage renal failure. But despite such treatment, about 60% of these patients die or develop permanent renal damage within 1 year.2,3,24
Eculizumab, a monoclonal antibody against complement component C5, has been approved by the US Food and Drug Administration for atypical hemolytic-uremic syndrome and may improve quality of life.25–27
PLASMA EXCHANGE IS THE MAINSTAY OF THERAPY
In 2012, the British Society for Haematology published revised guidelines for managing TTP and other thrombotic microangiopathies.28
Acquired idiopathic TTP with reduced ADAMTS13 activity requires immediate therapeutic plasma exchange. Daily plasma exchange combines plasmapheresis to remove circulating ultralarge von Willebrand factor-platelet strings and autoantibodies against ADAMTS13, and infusion of fresh-frozen plasma to replace ADAMTS13.18 This procedure is the mainstay of therapy and brings 70% to 90% of patients with idiopathic TTP to remission.1,2,5,6 However, the optimal duration of daily plasma exchange and the number of procedures required is highly variable according to clinical condition. Therapeutic plasma exchange can also cause plasma-related adverse reactions.9,28 Congenital TTP requires plasma infusion or exchange depending on the patient’s severity of ADAMTS13 deficiency.
Corticosteroids are used in combination with daily therapeutic plasma exchange, although evidence from controlled trials of their efficacy in this setting is lacking. Patients with severely decreased ADAMTS13 activity or low titers of ADAMTS13 autoantibodies tend to respond to the therapy.5,8,29
An ADAMTS13 assay with a short turn-around time can help guide the decision to initiate therapeutic plasma exchange. However, if there is a strong clinical suspicion of TTP, plasma exchange should be initiated immediately without waiting for test results.5,30 Monitoring ADAMTS13 activity or inhibitor during initial plasma exchange therapy has had conflicting results in several studies and is generally not recommended for patients with acquired TTP.8,30,31
RELAPSE IS COMMON
About 20% to 50% of patients with idiopathic TTP experience a relapse (Case 2). Most relapses occur within the first 2 years after the initial episode, with an estimated risk of 43% for relapse at 7.5 years.5,9
Factors that predict a higher risk of relapse include persistently severely decreased ADAMTS13 activity, positive inhibitor, and high titers of autoantibodies to ADAMTS13 during symptomatic TTP. During clinical remission, persistence of autoantibodies also indicates increased risk.1,3,5,6,9
Patients who have a relapse and whose disease is refractory to therapeutic plasma exchange (10%–20% of cases) have been treated with corticosteroids, splenectomy, or immunosuppressive agents (cyclosporine, azathioprine, or cyclophosphamide) with varying rates of success. Rituximab (monoclonal anti-CD20) has recently been used as second-line therapy in refractory or relapsing immune-mediated TTP or idiopathic TTP with neurologic or cardiac symptoms associated with a poor prognosis. Therapy including rituximab results in improved response and progression-free survival.32 Other potential therapies, including recombinant active ADAMTS13, are under investigation.9,23,28,30,33,34
- Sadler JE, Moake JL, Miyata T, George JN. Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2004; 1:407–423.
- Shenkman B, Einav Y. Thrombotic thrombocytopenic purpura and other thrombotic microangiopathic hemolytic anemias: diagnosis and classification. Autoimmun Rev 2014; 13:584–586.
- Shah N, Sarode R. Thrombotic thrombocytopenic purpura-what is new? J Clin Apher 2013; 28:30–35.
- Imanirad I, Rajasekhar A, Zumberg M. A case series of atypical presentations of thrombotic thrombocytopenic purpura. J Clin Apher 2012; 27:221–226.
- George JN, Al-Nouri ZL. Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes. Hematology Am Soc Hematol Educ Program 2012; 1:604–609.
- Shah N, Rutherford C, Matevosyan K, Shen YM, Sarode R. Role of ADAMTS13 in the management of thrombotic microangiopathies including thrombotic thrombocytopenic purpura (TTP). Br J Haematol 2013; 163:514–519.
- Cataland SR, Yang S, Wu HM. The use of ADAMTS13 activity, platelet count, and serum creatinine to differentiate acquired thrombotic thrombocytopenic purpura from other thrombotic microangiopathies. Br J Haematol 2012; 157:501–503.
- Mannucci PM, Peyvandi F. TTP and ADAMTS13: when Is testing appropriate? Hematology Am Soc Hematol Educ Program 2007; 1:121–126.
- Chaturved S, Carcioppolo D, Zhang L, McCar KR. Management and outcomes of patients with TTP: analysis of 100 cases at a single institution. Am J Hematol 2013; 88:560–565.
- Peyvandi F, Palla R, Lotta LA, Mackie I, Scully MA, Machin SJ. ADAMTS-13 assays in thrombotic thrombocytopenic purpura. J Thromb Haemost 2010; 8:631–640.
- Cataland SR, Scully MA, Paskavitz J, et al. Evidence of persistent neurologic injury following thrombotic thrombocytopenic purpura. Am J Hematol 2011; 86:87–89.
- Meloni G, Proia A, Antonini G, et al. Thrombotic thrombocytopenic purpura: prospective neurologic, neuroimaging and neurophysiologic evaluation. Haematologica 2001; 86:1194–1199.
- Kwaan HC, Boggio LN. The clinical spectrum of thrombotic thrombocytopenic purpura. Semin Thromb Hemost 2005; 31:673–680.
- Sarode R. Atypical presentations of thrombotic thrombocytopenic purpura: a review. J Clin Apher 2009; 24:47–52.
- Volcy J, Nzerue CM, Oderinde A, Hewan-Iowe K. Cocaine-induced acute renal failure, hemolysis, and thrombocytopenia mimicking thrombotic thrombocytopenic purpura. Am J Kidney Dis 2000; 35:E3.
- Kremer Hovinga JA, Mottini M, Lammle B. Measurement of ADAMTS-13 activity in plasma by the FRETS-VWF73 assay: comparison with other assay methods. J Thromb Haemost 2006; 4:1146–1148.
- Groot E, Hulstein JJ, Rison CN, de Groot PG, Fijnheer R. FRETS-VWF73: a rapid and predictive tool for thrombotic thrombocytopenic purpura. J Thromb Haemost 2006; 4:698–699.
- Barrows BD, Teruya J. Use of the ADAMTS13 activity assay improved the accuracy and efficiency of the diagnosis and treatment of suspected acquired thrombotic thrombocytopenic purpura. Arch Pathol Lab Med 2014; 138:546–549.
- Rieger M, Mannucci PM, Kremer Hovinga JA, et al. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood 2005; 106:1262–1267.
- Rogers HJ, Kottke-Marchant K. ADAMTS13 evaluation for thrombotic thrombocytopenic purpura. Pathology Innovations, Pathology and Laboratory Medicine Institute. Cleveland Clinic, Fall 2014:6–9.
- Józsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008; 111:1512–1514.
- Diamante Chiodini B, Davin JC, Corazza F, et al. Eculizumab in anti-factor H antibodies associated with atypical hemolytic uremic syndrome. Pediatrics 2014; 133:e1764–e1768.
- Taylor CM, Machin S, Wigmore SJ, Goodship TH; working party from the Renal Association, the British Committee for Standards in Haematology and the British Transplantation Society. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol 2009; 148:37–47.
- Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 2010; 36:673–681.
- Tsai HM, Kuo E. Eculizumab therapy leads to rapid resolution of thrombocytopenia in atypical hemolytic uremic syndrome. Adv Hematol 2014; 295323:1–7.
- Lapeyraque AL, Frémeaux-Bacchi V, Robitaille P. Efficacy of eculizumab in a patient with factor-H-associated atypical hemolytic uremic syndrome. Pediatr Nephrol 2011; 26:621–624.
- Baskin E, Gulleroglu K, Kantar A, Bayrakci U, Ozkaya O. Success of eculizumab in the treatment of atypical hemolytic uremic syndrome. Pediatr Nephrol 2015; 30:783–789.
- Scully M, Hunt BJ, Benjamin S, et al; British Committee for Standards in Haematology. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 2012; 158:323–325.
- Abassi E, Yawn D, Leveque E, Nolasco L, Lopez J, Moake J. Correlation of ADAMTS-13 activity with response to plasma exchange in patients diagnosed with thrombotic thrombocytopenic purpura (Abstract #3921). Blood 2004; 104:242a.
- Blombery P, Scully M. Management of thrombocytic thrombocytopenic purpura: current perspectives. J Blood Med 2014; 5:15–23.
- Wu N, Liu J, Yang S, et al. Diagnostic and prognostic values of ADAMTS13 activity measured during daily plasma exchange therapy in patients with acquired thrombotic thrombocytopenic purpura. Transfusion 2015; 55:18–24.
- Cuker A. Adjuvant rituximab to prevent TTP relapse. Blood 2016; 127:2952–2953.
- Chapman K, Yuen S. Therapy for thrombotic thrombocytopenic purpura: past, present and future. Semin Thromb Hemost 2014; 40:34–40.
- Heidel F, Lipka DB, von Auer C, Huber C, Schrarrer I, Hess G. Addition of rituximab to standard therapy improves response rate and progression-free survival in relapsed or refractory thrombotic thrombocytopenic purpura and autoimmune haemolytic anaemia. Thromb Haemost 2007; 97:228–233.
- Sadler JE, Moake JL, Miyata T, George JN. Recent advances in thrombotic thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2004; 1:407–423.
- Shenkman B, Einav Y. Thrombotic thrombocytopenic purpura and other thrombotic microangiopathic hemolytic anemias: diagnosis and classification. Autoimmun Rev 2014; 13:584–586.
- Shah N, Sarode R. Thrombotic thrombocytopenic purpura-what is new? J Clin Apher 2013; 28:30–35.
- Imanirad I, Rajasekhar A, Zumberg M. A case series of atypical presentations of thrombotic thrombocytopenic purpura. J Clin Apher 2012; 27:221–226.
- George JN, Al-Nouri ZL. Diagnostic and therapeutic challenges in the thrombotic thrombocytopenic purpura and hemolytic uremic syndromes. Hematology Am Soc Hematol Educ Program 2012; 1:604–609.
- Shah N, Rutherford C, Matevosyan K, Shen YM, Sarode R. Role of ADAMTS13 in the management of thrombotic microangiopathies including thrombotic thrombocytopenic purpura (TTP). Br J Haematol 2013; 163:514–519.
- Cataland SR, Yang S, Wu HM. The use of ADAMTS13 activity, platelet count, and serum creatinine to differentiate acquired thrombotic thrombocytopenic purpura from other thrombotic microangiopathies. Br J Haematol 2012; 157:501–503.
- Mannucci PM, Peyvandi F. TTP and ADAMTS13: when Is testing appropriate? Hematology Am Soc Hematol Educ Program 2007; 1:121–126.
- Chaturved S, Carcioppolo D, Zhang L, McCar KR. Management and outcomes of patients with TTP: analysis of 100 cases at a single institution. Am J Hematol 2013; 88:560–565.
- Peyvandi F, Palla R, Lotta LA, Mackie I, Scully MA, Machin SJ. ADAMTS-13 assays in thrombotic thrombocytopenic purpura. J Thromb Haemost 2010; 8:631–640.
- Cataland SR, Scully MA, Paskavitz J, et al. Evidence of persistent neurologic injury following thrombotic thrombocytopenic purpura. Am J Hematol 2011; 86:87–89.
- Meloni G, Proia A, Antonini G, et al. Thrombotic thrombocytopenic purpura: prospective neurologic, neuroimaging and neurophysiologic evaluation. Haematologica 2001; 86:1194–1199.
- Kwaan HC, Boggio LN. The clinical spectrum of thrombotic thrombocytopenic purpura. Semin Thromb Hemost 2005; 31:673–680.
- Sarode R. Atypical presentations of thrombotic thrombocytopenic purpura: a review. J Clin Apher 2009; 24:47–52.
- Volcy J, Nzerue CM, Oderinde A, Hewan-Iowe K. Cocaine-induced acute renal failure, hemolysis, and thrombocytopenia mimicking thrombotic thrombocytopenic purpura. Am J Kidney Dis 2000; 35:E3.
- Kremer Hovinga JA, Mottini M, Lammle B. Measurement of ADAMTS-13 activity in plasma by the FRETS-VWF73 assay: comparison with other assay methods. J Thromb Haemost 2006; 4:1146–1148.
- Groot E, Hulstein JJ, Rison CN, de Groot PG, Fijnheer R. FRETS-VWF73: a rapid and predictive tool for thrombotic thrombocytopenic purpura. J Thromb Haemost 2006; 4:698–699.
- Barrows BD, Teruya J. Use of the ADAMTS13 activity assay improved the accuracy and efficiency of the diagnosis and treatment of suspected acquired thrombotic thrombocytopenic purpura. Arch Pathol Lab Med 2014; 138:546–549.
- Rieger M, Mannucci PM, Kremer Hovinga JA, et al. ADAMTS13 autoantibodies in patients with thrombotic microangiopathies and other immunomediated diseases. Blood 2005; 106:1262–1267.
- Rogers HJ, Kottke-Marchant K. ADAMTS13 evaluation for thrombotic thrombocytopenic purpura. Pathology Innovations, Pathology and Laboratory Medicine Institute. Cleveland Clinic, Fall 2014:6–9.
- Józsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood 2008; 111:1512–1514.
- Diamante Chiodini B, Davin JC, Corazza F, et al. Eculizumab in anti-factor H antibodies associated with atypical hemolytic uremic syndrome. Pediatrics 2014; 133:e1764–e1768.
- Taylor CM, Machin S, Wigmore SJ, Goodship TH; working party from the Renal Association, the British Committee for Standards in Haematology and the British Transplantation Society. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol 2009; 148:37–47.
- Loirat C, Garnier A, Sellier-Leclerc AL, Kwon T. Plasmatherapy in atypical hemolytic uremic syndrome. Semin Thromb Hemost 2010; 36:673–681.
- Tsai HM, Kuo E. Eculizumab therapy leads to rapid resolution of thrombocytopenia in atypical hemolytic uremic syndrome. Adv Hematol 2014; 295323:1–7.
- Lapeyraque AL, Frémeaux-Bacchi V, Robitaille P. Efficacy of eculizumab in a patient with factor-H-associated atypical hemolytic uremic syndrome. Pediatr Nephrol 2011; 26:621–624.
- Baskin E, Gulleroglu K, Kantar A, Bayrakci U, Ozkaya O. Success of eculizumab in the treatment of atypical hemolytic uremic syndrome. Pediatr Nephrol 2015; 30:783–789.
- Scully M, Hunt BJ, Benjamin S, et al; British Committee for Standards in Haematology. Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Haematol 2012; 158:323–325.
- Abassi E, Yawn D, Leveque E, Nolasco L, Lopez J, Moake J. Correlation of ADAMTS-13 activity with response to plasma exchange in patients diagnosed with thrombotic thrombocytopenic purpura (Abstract #3921). Blood 2004; 104:242a.
- Blombery P, Scully M. Management of thrombocytic thrombocytopenic purpura: current perspectives. J Blood Med 2014; 5:15–23.
- Wu N, Liu J, Yang S, et al. Diagnostic and prognostic values of ADAMTS13 activity measured during daily plasma exchange therapy in patients with acquired thrombotic thrombocytopenic purpura. Transfusion 2015; 55:18–24.
- Cuker A. Adjuvant rituximab to prevent TTP relapse. Blood 2016; 127:2952–2953.
- Chapman K, Yuen S. Therapy for thrombotic thrombocytopenic purpura: past, present and future. Semin Thromb Hemost 2014; 40:34–40.
- Heidel F, Lipka DB, von Auer C, Huber C, Schrarrer I, Hess G. Addition of rituximab to standard therapy improves response rate and progression-free survival in relapsed or refractory thrombotic thrombocytopenic purpura and autoimmune haemolytic anaemia. Thromb Haemost 2007; 97:228–233.
KEY POINTS
- Symptoms of TTP are usually neurologic but can also be cardiac or abdominal. Thrombocytopenia and unexplained microangiopathic hemolytic anemia are sufficient to highly suspect the disease.
- In the appropriate clinical setting, an ADAMTS13 activity level lower than 10% is highly indicative of TTP.
- ADAMTS13 inhibitor and ADAMTS13 antibody assays provide more diagnostic clues. ADAMTS13 antibody is generally absent in the congenital form.
- The ADAMTS13 assay can help distinguish TTP from hemolytic-uremic syndrome, which presents similarly but typically involves normal or only mildly reduced ADAMTS13 activity.
- A strong clinical suspicion of TTP warrants immediate initiation of therapeutic plasma exchange without waiting for ADAMTS13 test results.