User login
A 34-year-old, previously healthy Japanese man developed a dry cough. He did not have dyspnea, nasal discharge, sore throat, facial pain, nasal congestion, or postnasal drip. His symptoms persisted despite several courses of antibiotics (from different physicians), including clarithromycin, minocycline, and levofloxacin. A chest x-ray after 2 months of symptoms and a noncontrast chest computed tomography (CT) after 4 months of symptoms were normal, and bacterial and mycobacterial sputum cultures were sterile. Treatment with salmeterol and fluticasone was ineffective.
The persistence of a cough for longer than 8 weeks constitutes chronic cough. The initial negative review of systems argues against several of the usual etiologies. The lack of nasal discharge, sore throat, facial pain, nasal congestion, and postnasal drip lessens the probability of upper airway cough syndrome. The absence of dyspnea decreases the likelihood of congestive heart failure, asthma, or chronic obstructive pulmonary disease. Additional history should include whether the patient has orthopnea, paroxysmal nocturnal dyspnea, or a reduced exercise tolerance.
The persistence of symptoms despite multiple courses of antibiotics suggests that the process is inflammatory but not infectious, that the infection is not susceptible to the selected antibiotics, that the antibiotics cannot penetrate the site of infection, or that the ongoing symptoms are related to the antibiotics themselves. Pathogens that may cause chronic cough for months include mycobacteria, fungi (eg, Aspergillus , endemic mycoses), and parasites (eg, Strongyloides , Paragonimus ). Even when appropriately treated, many infections may result in a prolonged cough (eg, pertussis). The fluoroquinolone and macrolide exposure may have suppressed the mycobacterial cultures. The lack of response to salmeterol and fluticasone lessens the probability of asthma.
After 4 months of symptoms, his cough worsened, and he developed dysphagia and odynophagia, particularly when he initiated swallowing. He experienced daily fevers with temperatures between 38.0°C and 38.5°C. A repeat chest x-ray was normal. His white blood cell count was 14,200 per μL, and the C-reactive protein (CRP) was 12.91 mg/dL (normal <0.24 mg/dL). His symptoms did not improve with additional courses of clarithromycin, levofloxacin, or moxifloxacin. After 5 months of symptoms, he was referred to the internal medicine clinic of a teaching hospital in Japan.
The patient’s fevers, leukocytosis, and elevated CRP signal an inflammatory process, but whether it is infectious or not remains uncertain. The normal repeat chest x-ray lessens the likelihood of a pulmonary infection. Difficulty with initiating a swallow characterizes oropharyngeal dysphagia which features coughing or choking with oral intake and is typically caused by neuromuscular conditions like stroke, amyotrophic lateral sclerosis, or myasthenia gravis. The coexistence of oropharyngeal dysphagia and odynophagia may indicate pharyngitis, a retropharyngeal or parapharyngeal abscess, or oropharyngeal cancer.
Esophageal dysphagia occurs several seconds following swallow initiation and may arise with mucosal, smooth muscle, or neuromuscular diseases of the esophagus. Concomitant dysphagia and odynophagia may indicate esophageal spasm or esophagitis. Causes of esophagitis include infection (eg, candidiasis, herpes simplex virus [HSV], cytomegalovirus [CMV], or human immunodeficiency virus [HIV]), infiltration (eg, eosinophilic esophagitis), or irritation (eg, from medication, caustic ingestion, or gastroesophageal reflux). He is at risk for esophageal candidiasis following multiple courses of antibiotics. Esophageal dysphagia occurring with liquids and solids may indicate disordered motility, as opposed to dysphagia with solids alone, which may signal endoluminal obstruction.
At his outpatient evaluation, he denied headache, vision changes, chest pain, hemoptysis, palpitations, abdominal pain, dysuria, musculoskeletal symptoms, anorexia, or symptoms of gastroesophageal reflux. He did not have chills, rigors, or night sweats, but he had lost 3.4 kg in 5 months. He had not traveled within or outside of Japan in many years and was not involved in outdoor activities. He was engaged to and monogamous with his female partner of 5 years. He smoked 10 cigarettes per day for 14 years but stopped smoking during the last 2 months on account of his symptoms. He drank 6 beers per month and worked as a researcher at a chemical company but did not have any inhalational exposures.
His weight loss could be from reduced caloric intake due to dysphagia and odynophagia or may reflect an energy deficit related to chronic illness and inflammatory state. His smoking history increases his risk of bronchopulmonary infection and malignancy. Bronchogenic carcinoma may present with chronic cough, fevers, weight loss, or dysphagia from external compression by lymphadenopathy or mediastinal disease; however, his young age and recent chest CT results make lung cancer unlikely.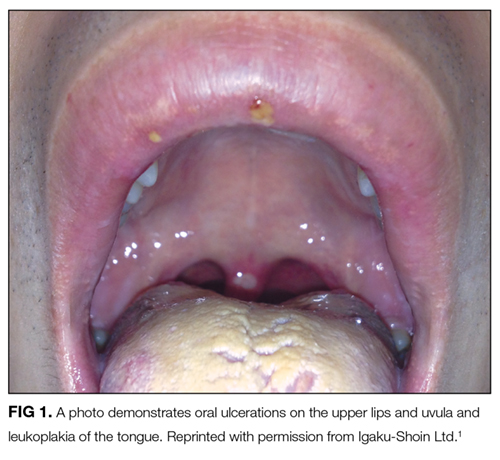
The white coating on his tongue could reflect oral leukoplakia, a reactive and potentially precancerous process that typically manifests as patches or plaques on oral mucosa. It can be distinguished from candidiasis, which scrapes off using a tongue blade. The extensive tongue coating is consistent with oral candidiasis. Potential predispositions include inhaled corticosteroids, antibiotic exposure, and/or an undiagnosed immunodeficiency syndrome (eg, HIV).
The initial diagnostic branch point for nontraumatic oral ulcers is infectious versus noninfectious. Infections that cause oral ulcers include HSV, CMV, and syphilis. The appearance and occurrence of the ulcers on freely moveable mucosa are consistent with aphthous stomatitis. Recurrent aphthous ulcers may occur in autoimmune diseases, including Behçet disease, Crohn disease, celiac sprue, and reactive arthritis. An endoscopy should be considered to detect esophageal ulcerations or esophageal candidiasis.
The rash may indicate folliculitis, usually attributable to Staphylococcus aureus or to Pseudomonas in the setting of recreational water exposure. Broad-spectrum antibiotics or immunodeficiency predisposes to candida folliculitis, while systemic candidiasis may cause metastatic skin lesions. The most common cutaneous manifestation of Behçet disease is erythema nodosum, but follicular and papulopustular lesions are also characteristic.

Pulmonary nodules are caused by infections, noninfectious inflammation, and malignancy. Infectious causes of pulmonary nodules include septic emboli, bacterial abscesses, and mycobacterial and fungal infection; noninfectious inflammatory causes include vasculitis (eg, granulomatosis with polyangiitis), rheumatoid arthritis, sarcoidosis, and lymphomatoid granulomatosis. Although additional culture data, serologic testing, and tuberculin skin testing or an interferon-gamma release assay may help to exclude these infections, the chronicity of symptoms, and lack of response to multiple antibiotic courses favor a noninfectious etiology.
Thickening of the aorta and left pulmonary artery may arise from an infectious, infiltrative, or inflammatory process. Arterial infections arise from direct inoculation, such as catheterization, trauma, or a contiguous site of infection, or from embolic seeding of atherosclerotic plaques or aneurysms. Malignant and nonmalignant processes, including sarcomas, lymphomas, histiocytoses (eg, Erdheim–Chester disease), and IgG4-related disease, may infiltrate the vascular walls. He has no evidence of visceral organ involvement to suggest these multisystem diagnoses.
The combined involvement of the aorta and pulmonary artery suggest a large-vessel vasculitis. Giant cell arteritis is exceedingly rare in patients younger than 50. Takayasu arteritis is a large-vessel vasculitis that predominantly affects women and may present with hypertension, arterial bruits, or discrepant blood pressure between arms, none of which were reported in this case. Behçet disease affects blood vessels of all sizes, including the aorta and pulmonary vasculature. His fevers, oral ulcers, perifollicular rash, and lymphadenopathy are consistent with this diagnosis, although he lacks the genital ulcers that occur in the majority of patients. Pulmonary nodules in Behçet disease arise from pulmonary or pleural vasculitis, resulting in focal inflammation, hemorrhage, or infarction. An ophthalmologic examination for uveitis and a pathergy test would support this diagnosis.
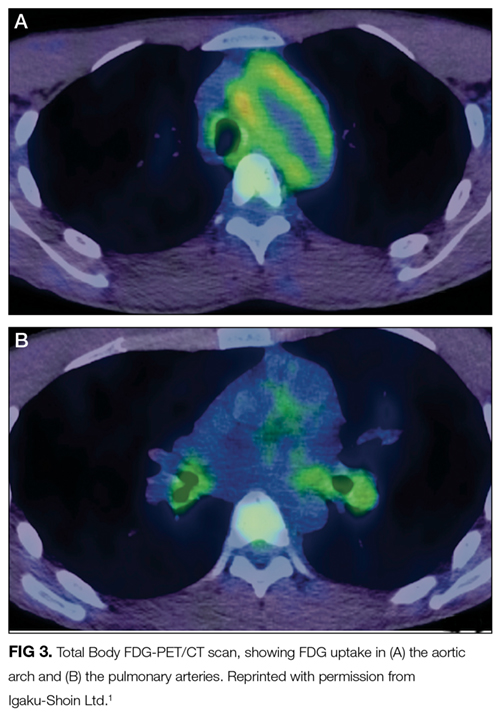
FDG accumulation in the aorta and pulmonary arteries signals large-vessel inflammation. The lack of FDG-avidity of the ground-glass opacities and nodular lesion suggests that these are not metabolically active tumors or infections but may be sequelae of the underlying disease, such as a hemorrhage or infarction from vasculitis. Sarcoidosis could account for the lung findings, but large-vessel vasculopathy would be exceedingly uncommon. Microscopic polyangiitis and granulomatosis with polyangiitis also cause pulmonary and vascular inflammation, but the nonreactive ANCA, absence of sinus disease, and normal urinalysis and kidney function make pauci-immune vasculitis unlikely. While the large-vessel involvement is consistent with Takayasu arteritis, the oral ulcers and rash are not.
Despite the absence of uveitis and the negative pathergy test, his oral aphthosis, papulopustular rash, and large-vessel vasculitis make Behçet disease the likely diagnosis. Behçet disease is most strongly associated with HLA B51, although other HLA haplotypes (including HLA A26 and HLA B52) are frequent in Behçet disease as well. As aortitis and pulmonary vasculitis can be associated with substantial morbidity and mortality, an urgent consultation with a rheumatologist regarding the initiation of immunosuppression is warranted.
Based on the mucocutaneous lesions, radiologic findings consistent with large-vessel vasculitis, and positive HLA A26 and HLA B52, he was diagnosed with Behçet disease. After 1 week of treatment with prednisolone 60 mg daily, his cough resolved and the oral aphthous ulcers and papulopustular rash improved. One month later, a chest CT showed significant reduction of the wall thickening of the aorta, its branches, and of the left pulmonary artery. The nodular lesion in the left lower lobe was unchanged, but the ground-glass opacities in the left upper lobe had disappeared.
When prednisolone was tapered down to 17.5 mg, his dry cough and low-grade fevers recurred, along with a slight elevation of inflammatory markers, and a ground-glass opacity appeared on the periphery of the left upper lobe. A sputum culture and fungal antigens were negative. His cough improved with the resumption of the previous dose of prednisolone. He remained symptom-free after 2 years of treatment with azathioprine 150 mg daily and prednisolone 2 mg daily and is now only treated with azathioprine.
DISCUSSION
Behçet disease is a multisystem vasculitis involving blood vessels of all sizes in the arterial and venous circulation that presents with oral and genital ulcers, ocular abnormalities (uveitis, retinitis), skin lesions (erythema nodosum, nonfollicular papulopustular lesions, or “pseudofolliculitis”), pathergy, and vascular lesions (thrombophlebitis, thrombosis, and aneurysm).
This patient presented with a chronic cough from pulmonary involvement by Behçet disease. The most common presenting symptom in a study of 47 patients with Behçet disease with pulmonary arteriopathy was hemoptysis followed by a nonbloody cough.2 Among these patients with pulmonary artery aneurysm, thrombosis, or both, 40 (85%) had nodules caused by infarction or inflammation and 21 (45%) had ground-glass opacities attributed to intraparenchymal hemorrhage. There are several case reports of chronic cough attributed to large-vessel vasculitis.3-5 Although the pathology of vasculitis-related cough is not fully understood, the inflammation of large vessels (aorta and pulmonary arteries) adjacent to the tracheobronchial tree may irritate regional cough receptors.3
Disease classification criteria are common in rheumatologic diseases; these criteria are developed to categorize patients for research studies and are not intended to diagnose individual patients.6 The classification criteria favor increased specificity at the expense of sensitivity to avoid misclassifying patients as having a disease, which would compromise the results of research studies. For instance, a study assessing a treatment for Behçet disease must exclude patients with inflammatory bowel disease, as these distinct patient populations may demonstrate discrepant responses to the investigative therapy. The specificity and homogeneity favored by classification criteria make those criteria inappropriate to rely on exclusively for the diagnosis of individual patients.7 The symptoms of many autoimmune diseases develop sequentially over time. Waiting for a patient with active, multisystem vasculitis to fulfill all of the Behçet disease classification criteria can lead to the harmful withholding of disease-modifying treatment.
The diagnosis of Behçet disease is made on clinical grounds; there is no gold standard test or histopathologic finding, and classification criteria remain imperfect. Although classification criteria help clinicians understand cardinal disease features, they cannot substitute for the more complex clinical reasoning required to establish a working diagnosis. The clinician must understand the pretest probability of disease, consider the presence or absence of characteristic features, exclude competing diagnoses, and decipher the risk-to-benefit ratio of therapeutic options and the urgency of treatment when assigning a diagnostic label. This patient’s pneumonitis, mucocutaneous changes, aortopathy, and compatible HLA typing (coupled with the exclusion of infectious diseases) were sufficient to diagnose Behçet disease. This case reminds us that classification criteria serve as a starting point, not as an end point, and that clinicians must ultimately make diagnoses and initiate treatment by thinking outside the checkbox.
TEACHING POINTS
- Large-vessel vasculitis is a rare cause of chronic cough.
- Although the most well-recognized signs of Behçet disease include genital and oral ulcers and uveitis, patients may also present with less common manifestations such as skin lesions (erythema nodosum, nonfollicular papulopustular lesions, or “pseudofolliculitis”) and vascular lesions of the artery (arteritis and aneurysm) and veins (thrombophlebitis and thrombosis).
- Classification criteria capture cardinal features of a disease but favor specificity over sensitivity and should not serve as a checklist for diagnosing a patient.
Acknowledgment
A brief version of this case was published as a case report in the Journal of Integrated Medicine 2013;23(12):1014-1017. Images from that publication were republished here with the permission of the publisher (Igaku-Shoin Ltd).
Disclosure
Dr. Dhaliwal reports receiving honoraria from ISMIE Mutual Insurance Company and Physicians’ Reciprocal Insurers. All other authors have nothing to disclose.
1. Kanamori M, Kubo T, Sakemi H. What’s your diagnosis? [in Japanese] J Integrated Med. 2013; 23 (12):1014-1017.
2. Seyahi E, Melikoglu M, Akman C, et al. Pulmonary artery involvement and associated lung disease in Behçet disease: a series of 47 patients. Medicine (Baltimore). 2012;91(1):35-48. PubMed
3. Olopade CO, Sekosan M, Schraufnagel DE. Giant cell arteritis manifesting as chronic cough and fever of unknown origin. Mayo Clin Proc. 1997;72(11):1048-1050. PubMed
4. Hellmann DB. Temporal arteritis: a cough, toothache, and tongue infarction. JAMA. 2002;287(22):2996-3000. PubMed
5. Karagiannis A, Mathiopoulou L, Tziomalos K, et al. Dry cough as first manifestation of giant-cell arteritis. J Am Geriatr Soc. 2006;54(12):1957-1958. PubMed
6. Aggarwal R, Ringold S, Khanna D, et al. Distinctions between diagnostic and classification criteria? Arthritis Care Res (Hoboken). 2015;67(7):891-897. PubMed
7. Rao JK, Allen NB, Pincus T. Limitations of the 1990 American College of Rheumatology classification criteria in the diagnosis of vasculitis. Ann Intern Med. 1998;129(5):345-352. PubMed
8. Davatchi F, Sadeghi Abdollahi B, Shahram F, Chams-Davatchi C, Shams H, Nadji A. Classification and Diagnosis Criteria for Behçet’s Disease. In: Emmi L, ed. Behçet’s Syndrome. From Pathogenesis to Treatment. Milan, Italy: Springer; 2014:189-198.
9. Criteria for diagnosis of Behcet’s disease. International Study Group for Behçet’s Disease. Lancet. 1990;335(8697):1078-1080. PubMed
10. Davatchi F, Assaad-Khalil S, Calamia KT, et al. The International Criteria for Behçet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338–347. PubMed
11. Suzuki Kurokawa M, Suzuki N. Behçet’s disease. Clin Exp Med. 2004;4(1):10-20. PubMed
A 34-year-old, previously healthy Japanese man developed a dry cough. He did not have dyspnea, nasal discharge, sore throat, facial pain, nasal congestion, or postnasal drip. His symptoms persisted despite several courses of antibiotics (from different physicians), including clarithromycin, minocycline, and levofloxacin. A chest x-ray after 2 months of symptoms and a noncontrast chest computed tomography (CT) after 4 months of symptoms were normal, and bacterial and mycobacterial sputum cultures were sterile. Treatment with salmeterol and fluticasone was ineffective.
The persistence of a cough for longer than 8 weeks constitutes chronic cough. The initial negative review of systems argues against several of the usual etiologies. The lack of nasal discharge, sore throat, facial pain, nasal congestion, and postnasal drip lessens the probability of upper airway cough syndrome. The absence of dyspnea decreases the likelihood of congestive heart failure, asthma, or chronic obstructive pulmonary disease. Additional history should include whether the patient has orthopnea, paroxysmal nocturnal dyspnea, or a reduced exercise tolerance.
The persistence of symptoms despite multiple courses of antibiotics suggests that the process is inflammatory but not infectious, that the infection is not susceptible to the selected antibiotics, that the antibiotics cannot penetrate the site of infection, or that the ongoing symptoms are related to the antibiotics themselves. Pathogens that may cause chronic cough for months include mycobacteria, fungi (eg, Aspergillus , endemic mycoses), and parasites (eg, Strongyloides , Paragonimus ). Even when appropriately treated, many infections may result in a prolonged cough (eg, pertussis). The fluoroquinolone and macrolide exposure may have suppressed the mycobacterial cultures. The lack of response to salmeterol and fluticasone lessens the probability of asthma.
After 4 months of symptoms, his cough worsened, and he developed dysphagia and odynophagia, particularly when he initiated swallowing. He experienced daily fevers with temperatures between 38.0°C and 38.5°C. A repeat chest x-ray was normal. His white blood cell count was 14,200 per μL, and the C-reactive protein (CRP) was 12.91 mg/dL (normal <0.24 mg/dL). His symptoms did not improve with additional courses of clarithromycin, levofloxacin, or moxifloxacin. After 5 months of symptoms, he was referred to the internal medicine clinic of a teaching hospital in Japan.
The patient’s fevers, leukocytosis, and elevated CRP signal an inflammatory process, but whether it is infectious or not remains uncertain. The normal repeat chest x-ray lessens the likelihood of a pulmonary infection. Difficulty with initiating a swallow characterizes oropharyngeal dysphagia which features coughing or choking with oral intake and is typically caused by neuromuscular conditions like stroke, amyotrophic lateral sclerosis, or myasthenia gravis. The coexistence of oropharyngeal dysphagia and odynophagia may indicate pharyngitis, a retropharyngeal or parapharyngeal abscess, or oropharyngeal cancer.
Esophageal dysphagia occurs several seconds following swallow initiation and may arise with mucosal, smooth muscle, or neuromuscular diseases of the esophagus. Concomitant dysphagia and odynophagia may indicate esophageal spasm or esophagitis. Causes of esophagitis include infection (eg, candidiasis, herpes simplex virus [HSV], cytomegalovirus [CMV], or human immunodeficiency virus [HIV]), infiltration (eg, eosinophilic esophagitis), or irritation (eg, from medication, caustic ingestion, or gastroesophageal reflux). He is at risk for esophageal candidiasis following multiple courses of antibiotics. Esophageal dysphagia occurring with liquids and solids may indicate disordered motility, as opposed to dysphagia with solids alone, which may signal endoluminal obstruction.
At his outpatient evaluation, he denied headache, vision changes, chest pain, hemoptysis, palpitations, abdominal pain, dysuria, musculoskeletal symptoms, anorexia, or symptoms of gastroesophageal reflux. He did not have chills, rigors, or night sweats, but he had lost 3.4 kg in 5 months. He had not traveled within or outside of Japan in many years and was not involved in outdoor activities. He was engaged to and monogamous with his female partner of 5 years. He smoked 10 cigarettes per day for 14 years but stopped smoking during the last 2 months on account of his symptoms. He drank 6 beers per month and worked as a researcher at a chemical company but did not have any inhalational exposures.
His weight loss could be from reduced caloric intake due to dysphagia and odynophagia or may reflect an energy deficit related to chronic illness and inflammatory state. His smoking history increases his risk of bronchopulmonary infection and malignancy. Bronchogenic carcinoma may present with chronic cough, fevers, weight loss, or dysphagia from external compression by lymphadenopathy or mediastinal disease; however, his young age and recent chest CT results make lung cancer unlikely.
The white coating on his tongue could reflect oral leukoplakia, a reactive and potentially precancerous process that typically manifests as patches or plaques on oral mucosa. It can be distinguished from candidiasis, which scrapes off using a tongue blade. The extensive tongue coating is consistent with oral candidiasis. Potential predispositions include inhaled corticosteroids, antibiotic exposure, and/or an undiagnosed immunodeficiency syndrome (eg, HIV).
The initial diagnostic branch point for nontraumatic oral ulcers is infectious versus noninfectious. Infections that cause oral ulcers include HSV, CMV, and syphilis. The appearance and occurrence of the ulcers on freely moveable mucosa are consistent with aphthous stomatitis. Recurrent aphthous ulcers may occur in autoimmune diseases, including Behçet disease, Crohn disease, celiac sprue, and reactive arthritis. An endoscopy should be considered to detect esophageal ulcerations or esophageal candidiasis.
The rash may indicate folliculitis, usually attributable to Staphylococcus aureus or to Pseudomonas in the setting of recreational water exposure. Broad-spectrum antibiotics or immunodeficiency predisposes to candida folliculitis, while systemic candidiasis may cause metastatic skin lesions. The most common cutaneous manifestation of Behçet disease is erythema nodosum, but follicular and papulopustular lesions are also characteristic.
Pulmonary nodules are caused by infections, noninfectious inflammation, and malignancy. Infectious causes of pulmonary nodules include septic emboli, bacterial abscesses, and mycobacterial and fungal infection; noninfectious inflammatory causes include vasculitis (eg, granulomatosis with polyangiitis), rheumatoid arthritis, sarcoidosis, and lymphomatoid granulomatosis. Although additional culture data, serologic testing, and tuberculin skin testing or an interferon-gamma release assay may help to exclude these infections, the chronicity of symptoms, and lack of response to multiple antibiotic courses favor a noninfectious etiology.
Thickening of the aorta and left pulmonary artery may arise from an infectious, infiltrative, or inflammatory process. Arterial infections arise from direct inoculation, such as catheterization, trauma, or a contiguous site of infection, or from embolic seeding of atherosclerotic plaques or aneurysms. Malignant and nonmalignant processes, including sarcomas, lymphomas, histiocytoses (eg, Erdheim–Chester disease), and IgG4-related disease, may infiltrate the vascular walls. He has no evidence of visceral organ involvement to suggest these multisystem diagnoses.
The combined involvement of the aorta and pulmonary artery suggest a large-vessel vasculitis. Giant cell arteritis is exceedingly rare in patients younger than 50. Takayasu arteritis is a large-vessel vasculitis that predominantly affects women and may present with hypertension, arterial bruits, or discrepant blood pressure between arms, none of which were reported in this case. Behçet disease affects blood vessels of all sizes, including the aorta and pulmonary vasculature. His fevers, oral ulcers, perifollicular rash, and lymphadenopathy are consistent with this diagnosis, although he lacks the genital ulcers that occur in the majority of patients. Pulmonary nodules in Behçet disease arise from pulmonary or pleural vasculitis, resulting in focal inflammation, hemorrhage, or infarction. An ophthalmologic examination for uveitis and a pathergy test would support this diagnosis.
FDG accumulation in the aorta and pulmonary arteries signals large-vessel inflammation. The lack of FDG-avidity of the ground-glass opacities and nodular lesion suggests that these are not metabolically active tumors or infections but may be sequelae of the underlying disease, such as a hemorrhage or infarction from vasculitis. Sarcoidosis could account for the lung findings, but large-vessel vasculopathy would be exceedingly uncommon. Microscopic polyangiitis and granulomatosis with polyangiitis also cause pulmonary and vascular inflammation, but the nonreactive ANCA, absence of sinus disease, and normal urinalysis and kidney function make pauci-immune vasculitis unlikely. While the large-vessel involvement is consistent with Takayasu arteritis, the oral ulcers and rash are not.
Despite the absence of uveitis and the negative pathergy test, his oral aphthosis, papulopustular rash, and large-vessel vasculitis make Behçet disease the likely diagnosis. Behçet disease is most strongly associated with HLA B51, although other HLA haplotypes (including HLA A26 and HLA B52) are frequent in Behçet disease as well. As aortitis and pulmonary vasculitis can be associated with substantial morbidity and mortality, an urgent consultation with a rheumatologist regarding the initiation of immunosuppression is warranted.
Based on the mucocutaneous lesions, radiologic findings consistent with large-vessel vasculitis, and positive HLA A26 and HLA B52, he was diagnosed with Behçet disease. After 1 week of treatment with prednisolone 60 mg daily, his cough resolved and the oral aphthous ulcers and papulopustular rash improved. One month later, a chest CT showed significant reduction of the wall thickening of the aorta, its branches, and of the left pulmonary artery. The nodular lesion in the left lower lobe was unchanged, but the ground-glass opacities in the left upper lobe had disappeared.
When prednisolone was tapered down to 17.5 mg, his dry cough and low-grade fevers recurred, along with a slight elevation of inflammatory markers, and a ground-glass opacity appeared on the periphery of the left upper lobe. A sputum culture and fungal antigens were negative. His cough improved with the resumption of the previous dose of prednisolone. He remained symptom-free after 2 years of treatment with azathioprine 150 mg daily and prednisolone 2 mg daily and is now only treated with azathioprine.
DISCUSSION
Behçet disease is a multisystem vasculitis involving blood vessels of all sizes in the arterial and venous circulation that presents with oral and genital ulcers, ocular abnormalities (uveitis, retinitis), skin lesions (erythema nodosum, nonfollicular papulopustular lesions, or “pseudofolliculitis”), pathergy, and vascular lesions (thrombophlebitis, thrombosis, and aneurysm).
This patient presented with a chronic cough from pulmonary involvement by Behçet disease. The most common presenting symptom in a study of 47 patients with Behçet disease with pulmonary arteriopathy was hemoptysis followed by a nonbloody cough.2 Among these patients with pulmonary artery aneurysm, thrombosis, or both, 40 (85%) had nodules caused by infarction or inflammation and 21 (45%) had ground-glass opacities attributed to intraparenchymal hemorrhage. There are several case reports of chronic cough attributed to large-vessel vasculitis.3-5 Although the pathology of vasculitis-related cough is not fully understood, the inflammation of large vessels (aorta and pulmonary arteries) adjacent to the tracheobronchial tree may irritate regional cough receptors.3
Disease classification criteria are common in rheumatologic diseases; these criteria are developed to categorize patients for research studies and are not intended to diagnose individual patients.6 The classification criteria favor increased specificity at the expense of sensitivity to avoid misclassifying patients as having a disease, which would compromise the results of research studies. For instance, a study assessing a treatment for Behçet disease must exclude patients with inflammatory bowel disease, as these distinct patient populations may demonstrate discrepant responses to the investigative therapy. The specificity and homogeneity favored by classification criteria make those criteria inappropriate to rely on exclusively for the diagnosis of individual patients.7 The symptoms of many autoimmune diseases develop sequentially over time. Waiting for a patient with active, multisystem vasculitis to fulfill all of the Behçet disease classification criteria can lead to the harmful withholding of disease-modifying treatment.
The diagnosis of Behçet disease is made on clinical grounds; there is no gold standard test or histopathologic finding, and classification criteria remain imperfect. Although classification criteria help clinicians understand cardinal disease features, they cannot substitute for the more complex clinical reasoning required to establish a working diagnosis. The clinician must understand the pretest probability of disease, consider the presence or absence of characteristic features, exclude competing diagnoses, and decipher the risk-to-benefit ratio of therapeutic options and the urgency of treatment when assigning a diagnostic label. This patient’s pneumonitis, mucocutaneous changes, aortopathy, and compatible HLA typing (coupled with the exclusion of infectious diseases) were sufficient to diagnose Behçet disease. This case reminds us that classification criteria serve as a starting point, not as an end point, and that clinicians must ultimately make diagnoses and initiate treatment by thinking outside the checkbox.
TEACHING POINTS
- Large-vessel vasculitis is a rare cause of chronic cough.
- Although the most well-recognized signs of Behçet disease include genital and oral ulcers and uveitis, patients may also present with less common manifestations such as skin lesions (erythema nodosum, nonfollicular papulopustular lesions, or “pseudofolliculitis”) and vascular lesions of the artery (arteritis and aneurysm) and veins (thrombophlebitis and thrombosis).
- Classification criteria capture cardinal features of a disease but favor specificity over sensitivity and should not serve as a checklist for diagnosing a patient.
Acknowledgment
A brief version of this case was published as a case report in the Journal of Integrated Medicine 2013;23(12):1014-1017. Images from that publication were republished here with the permission of the publisher (Igaku-Shoin Ltd).
Disclosure
Dr. Dhaliwal reports receiving honoraria from ISMIE Mutual Insurance Company and Physicians’ Reciprocal Insurers. All other authors have nothing to disclose.
A 34-year-old, previously healthy Japanese man developed a dry cough. He did not have dyspnea, nasal discharge, sore throat, facial pain, nasal congestion, or postnasal drip. His symptoms persisted despite several courses of antibiotics (from different physicians), including clarithromycin, minocycline, and levofloxacin. A chest x-ray after 2 months of symptoms and a noncontrast chest computed tomography (CT) after 4 months of symptoms were normal, and bacterial and mycobacterial sputum cultures were sterile. Treatment with salmeterol and fluticasone was ineffective.
The persistence of a cough for longer than 8 weeks constitutes chronic cough. The initial negative review of systems argues against several of the usual etiologies. The lack of nasal discharge, sore throat, facial pain, nasal congestion, and postnasal drip lessens the probability of upper airway cough syndrome. The absence of dyspnea decreases the likelihood of congestive heart failure, asthma, or chronic obstructive pulmonary disease. Additional history should include whether the patient has orthopnea, paroxysmal nocturnal dyspnea, or a reduced exercise tolerance.
The persistence of symptoms despite multiple courses of antibiotics suggests that the process is inflammatory but not infectious, that the infection is not susceptible to the selected antibiotics, that the antibiotics cannot penetrate the site of infection, or that the ongoing symptoms are related to the antibiotics themselves. Pathogens that may cause chronic cough for months include mycobacteria, fungi (eg, Aspergillus , endemic mycoses), and parasites (eg, Strongyloides , Paragonimus ). Even when appropriately treated, many infections may result in a prolonged cough (eg, pertussis). The fluoroquinolone and macrolide exposure may have suppressed the mycobacterial cultures. The lack of response to salmeterol and fluticasone lessens the probability of asthma.
After 4 months of symptoms, his cough worsened, and he developed dysphagia and odynophagia, particularly when he initiated swallowing. He experienced daily fevers with temperatures between 38.0°C and 38.5°C. A repeat chest x-ray was normal. His white blood cell count was 14,200 per μL, and the C-reactive protein (CRP) was 12.91 mg/dL (normal <0.24 mg/dL). His symptoms did not improve with additional courses of clarithromycin, levofloxacin, or moxifloxacin. After 5 months of symptoms, he was referred to the internal medicine clinic of a teaching hospital in Japan.
The patient’s fevers, leukocytosis, and elevated CRP signal an inflammatory process, but whether it is infectious or not remains uncertain. The normal repeat chest x-ray lessens the likelihood of a pulmonary infection. Difficulty with initiating a swallow characterizes oropharyngeal dysphagia which features coughing or choking with oral intake and is typically caused by neuromuscular conditions like stroke, amyotrophic lateral sclerosis, or myasthenia gravis. The coexistence of oropharyngeal dysphagia and odynophagia may indicate pharyngitis, a retropharyngeal or parapharyngeal abscess, or oropharyngeal cancer.
Esophageal dysphagia occurs several seconds following swallow initiation and may arise with mucosal, smooth muscle, or neuromuscular diseases of the esophagus. Concomitant dysphagia and odynophagia may indicate esophageal spasm or esophagitis. Causes of esophagitis include infection (eg, candidiasis, herpes simplex virus [HSV], cytomegalovirus [CMV], or human immunodeficiency virus [HIV]), infiltration (eg, eosinophilic esophagitis), or irritation (eg, from medication, caustic ingestion, or gastroesophageal reflux). He is at risk for esophageal candidiasis following multiple courses of antibiotics. Esophageal dysphagia occurring with liquids and solids may indicate disordered motility, as opposed to dysphagia with solids alone, which may signal endoluminal obstruction.
At his outpatient evaluation, he denied headache, vision changes, chest pain, hemoptysis, palpitations, abdominal pain, dysuria, musculoskeletal symptoms, anorexia, or symptoms of gastroesophageal reflux. He did not have chills, rigors, or night sweats, but he had lost 3.4 kg in 5 months. He had not traveled within or outside of Japan in many years and was not involved in outdoor activities. He was engaged to and monogamous with his female partner of 5 years. He smoked 10 cigarettes per day for 14 years but stopped smoking during the last 2 months on account of his symptoms. He drank 6 beers per month and worked as a researcher at a chemical company but did not have any inhalational exposures.
His weight loss could be from reduced caloric intake due to dysphagia and odynophagia or may reflect an energy deficit related to chronic illness and inflammatory state. His smoking history increases his risk of bronchopulmonary infection and malignancy. Bronchogenic carcinoma may present with chronic cough, fevers, weight loss, or dysphagia from external compression by lymphadenopathy or mediastinal disease; however, his young age and recent chest CT results make lung cancer unlikely.
The white coating on his tongue could reflect oral leukoplakia, a reactive and potentially precancerous process that typically manifests as patches or plaques on oral mucosa. It can be distinguished from candidiasis, which scrapes off using a tongue blade. The extensive tongue coating is consistent with oral candidiasis. Potential predispositions include inhaled corticosteroids, antibiotic exposure, and/or an undiagnosed immunodeficiency syndrome (eg, HIV).
The initial diagnostic branch point for nontraumatic oral ulcers is infectious versus noninfectious. Infections that cause oral ulcers include HSV, CMV, and syphilis. The appearance and occurrence of the ulcers on freely moveable mucosa are consistent with aphthous stomatitis. Recurrent aphthous ulcers may occur in autoimmune diseases, including Behçet disease, Crohn disease, celiac sprue, and reactive arthritis. An endoscopy should be considered to detect esophageal ulcerations or esophageal candidiasis.
The rash may indicate folliculitis, usually attributable to Staphylococcus aureus or to Pseudomonas in the setting of recreational water exposure. Broad-spectrum antibiotics or immunodeficiency predisposes to candida folliculitis, while systemic candidiasis may cause metastatic skin lesions. The most common cutaneous manifestation of Behçet disease is erythema nodosum, but follicular and papulopustular lesions are also characteristic.
Pulmonary nodules are caused by infections, noninfectious inflammation, and malignancy. Infectious causes of pulmonary nodules include septic emboli, bacterial abscesses, and mycobacterial and fungal infection; noninfectious inflammatory causes include vasculitis (eg, granulomatosis with polyangiitis), rheumatoid arthritis, sarcoidosis, and lymphomatoid granulomatosis. Although additional culture data, serologic testing, and tuberculin skin testing or an interferon-gamma release assay may help to exclude these infections, the chronicity of symptoms, and lack of response to multiple antibiotic courses favor a noninfectious etiology.
Thickening of the aorta and left pulmonary artery may arise from an infectious, infiltrative, or inflammatory process. Arterial infections arise from direct inoculation, such as catheterization, trauma, or a contiguous site of infection, or from embolic seeding of atherosclerotic plaques or aneurysms. Malignant and nonmalignant processes, including sarcomas, lymphomas, histiocytoses (eg, Erdheim–Chester disease), and IgG4-related disease, may infiltrate the vascular walls. He has no evidence of visceral organ involvement to suggest these multisystem diagnoses.
The combined involvement of the aorta and pulmonary artery suggest a large-vessel vasculitis. Giant cell arteritis is exceedingly rare in patients younger than 50. Takayasu arteritis is a large-vessel vasculitis that predominantly affects women and may present with hypertension, arterial bruits, or discrepant blood pressure between arms, none of which were reported in this case. Behçet disease affects blood vessels of all sizes, including the aorta and pulmonary vasculature. His fevers, oral ulcers, perifollicular rash, and lymphadenopathy are consistent with this diagnosis, although he lacks the genital ulcers that occur in the majority of patients. Pulmonary nodules in Behçet disease arise from pulmonary or pleural vasculitis, resulting in focal inflammation, hemorrhage, or infarction. An ophthalmologic examination for uveitis and a pathergy test would support this diagnosis.
FDG accumulation in the aorta and pulmonary arteries signals large-vessel inflammation. The lack of FDG-avidity of the ground-glass opacities and nodular lesion suggests that these are not metabolically active tumors or infections but may be sequelae of the underlying disease, such as a hemorrhage or infarction from vasculitis. Sarcoidosis could account for the lung findings, but large-vessel vasculopathy would be exceedingly uncommon. Microscopic polyangiitis and granulomatosis with polyangiitis also cause pulmonary and vascular inflammation, but the nonreactive ANCA, absence of sinus disease, and normal urinalysis and kidney function make pauci-immune vasculitis unlikely. While the large-vessel involvement is consistent with Takayasu arteritis, the oral ulcers and rash are not.
Despite the absence of uveitis and the negative pathergy test, his oral aphthosis, papulopustular rash, and large-vessel vasculitis make Behçet disease the likely diagnosis. Behçet disease is most strongly associated with HLA B51, although other HLA haplotypes (including HLA A26 and HLA B52) are frequent in Behçet disease as well. As aortitis and pulmonary vasculitis can be associated with substantial morbidity and mortality, an urgent consultation with a rheumatologist regarding the initiation of immunosuppression is warranted.
Based on the mucocutaneous lesions, radiologic findings consistent with large-vessel vasculitis, and positive HLA A26 and HLA B52, he was diagnosed with Behçet disease. After 1 week of treatment with prednisolone 60 mg daily, his cough resolved and the oral aphthous ulcers and papulopustular rash improved. One month later, a chest CT showed significant reduction of the wall thickening of the aorta, its branches, and of the left pulmonary artery. The nodular lesion in the left lower lobe was unchanged, but the ground-glass opacities in the left upper lobe had disappeared.
When prednisolone was tapered down to 17.5 mg, his dry cough and low-grade fevers recurred, along with a slight elevation of inflammatory markers, and a ground-glass opacity appeared on the periphery of the left upper lobe. A sputum culture and fungal antigens were negative. His cough improved with the resumption of the previous dose of prednisolone. He remained symptom-free after 2 years of treatment with azathioprine 150 mg daily and prednisolone 2 mg daily and is now only treated with azathioprine.
DISCUSSION
Behçet disease is a multisystem vasculitis involving blood vessels of all sizes in the arterial and venous circulation that presents with oral and genital ulcers, ocular abnormalities (uveitis, retinitis), skin lesions (erythema nodosum, nonfollicular papulopustular lesions, or “pseudofolliculitis”), pathergy, and vascular lesions (thrombophlebitis, thrombosis, and aneurysm).
This patient presented with a chronic cough from pulmonary involvement by Behçet disease. The most common presenting symptom in a study of 47 patients with Behçet disease with pulmonary arteriopathy was hemoptysis followed by a nonbloody cough.2 Among these patients with pulmonary artery aneurysm, thrombosis, or both, 40 (85%) had nodules caused by infarction or inflammation and 21 (45%) had ground-glass opacities attributed to intraparenchymal hemorrhage. There are several case reports of chronic cough attributed to large-vessel vasculitis.3-5 Although the pathology of vasculitis-related cough is not fully understood, the inflammation of large vessels (aorta and pulmonary arteries) adjacent to the tracheobronchial tree may irritate regional cough receptors.3
Disease classification criteria are common in rheumatologic diseases; these criteria are developed to categorize patients for research studies and are not intended to diagnose individual patients.6 The classification criteria favor increased specificity at the expense of sensitivity to avoid misclassifying patients as having a disease, which would compromise the results of research studies. For instance, a study assessing a treatment for Behçet disease must exclude patients with inflammatory bowel disease, as these distinct patient populations may demonstrate discrepant responses to the investigative therapy. The specificity and homogeneity favored by classification criteria make those criteria inappropriate to rely on exclusively for the diagnosis of individual patients.7 The symptoms of many autoimmune diseases develop sequentially over time. Waiting for a patient with active, multisystem vasculitis to fulfill all of the Behçet disease classification criteria can lead to the harmful withholding of disease-modifying treatment.
The diagnosis of Behçet disease is made on clinical grounds; there is no gold standard test or histopathologic finding, and classification criteria remain imperfect. Although classification criteria help clinicians understand cardinal disease features, they cannot substitute for the more complex clinical reasoning required to establish a working diagnosis. The clinician must understand the pretest probability of disease, consider the presence or absence of characteristic features, exclude competing diagnoses, and decipher the risk-to-benefit ratio of therapeutic options and the urgency of treatment when assigning a diagnostic label. This patient’s pneumonitis, mucocutaneous changes, aortopathy, and compatible HLA typing (coupled with the exclusion of infectious diseases) were sufficient to diagnose Behçet disease. This case reminds us that classification criteria serve as a starting point, not as an end point, and that clinicians must ultimately make diagnoses and initiate treatment by thinking outside the checkbox.
TEACHING POINTS
- Large-vessel vasculitis is a rare cause of chronic cough.
- Although the most well-recognized signs of Behçet disease include genital and oral ulcers and uveitis, patients may also present with less common manifestations such as skin lesions (erythema nodosum, nonfollicular papulopustular lesions, or “pseudofolliculitis”) and vascular lesions of the artery (arteritis and aneurysm) and veins (thrombophlebitis and thrombosis).
- Classification criteria capture cardinal features of a disease but favor specificity over sensitivity and should not serve as a checklist for diagnosing a patient.
Acknowledgment
A brief version of this case was published as a case report in the Journal of Integrated Medicine 2013;23(12):1014-1017. Images from that publication were republished here with the permission of the publisher (Igaku-Shoin Ltd).
Disclosure
Dr. Dhaliwal reports receiving honoraria from ISMIE Mutual Insurance Company and Physicians’ Reciprocal Insurers. All other authors have nothing to disclose.
1. Kanamori M, Kubo T, Sakemi H. What’s your diagnosis? [in Japanese] J Integrated Med. 2013; 23 (12):1014-1017.
2. Seyahi E, Melikoglu M, Akman C, et al. Pulmonary artery involvement and associated lung disease in Behçet disease: a series of 47 patients. Medicine (Baltimore). 2012;91(1):35-48. PubMed
3. Olopade CO, Sekosan M, Schraufnagel DE. Giant cell arteritis manifesting as chronic cough and fever of unknown origin. Mayo Clin Proc. 1997;72(11):1048-1050. PubMed
4. Hellmann DB. Temporal arteritis: a cough, toothache, and tongue infarction. JAMA. 2002;287(22):2996-3000. PubMed
5. Karagiannis A, Mathiopoulou L, Tziomalos K, et al. Dry cough as first manifestation of giant-cell arteritis. J Am Geriatr Soc. 2006;54(12):1957-1958. PubMed
6. Aggarwal R, Ringold S, Khanna D, et al. Distinctions between diagnostic and classification criteria? Arthritis Care Res (Hoboken). 2015;67(7):891-897. PubMed
7. Rao JK, Allen NB, Pincus T. Limitations of the 1990 American College of Rheumatology classification criteria in the diagnosis of vasculitis. Ann Intern Med. 1998;129(5):345-352. PubMed
8. Davatchi F, Sadeghi Abdollahi B, Shahram F, Chams-Davatchi C, Shams H, Nadji A. Classification and Diagnosis Criteria for Behçet’s Disease. In: Emmi L, ed. Behçet’s Syndrome. From Pathogenesis to Treatment. Milan, Italy: Springer; 2014:189-198.
9. Criteria for diagnosis of Behcet’s disease. International Study Group for Behçet’s Disease. Lancet. 1990;335(8697):1078-1080. PubMed
10. Davatchi F, Assaad-Khalil S, Calamia KT, et al. The International Criteria for Behçet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338–347. PubMed
11. Suzuki Kurokawa M, Suzuki N. Behçet’s disease. Clin Exp Med. 2004;4(1):10-20. PubMed
1. Kanamori M, Kubo T, Sakemi H. What’s your diagnosis? [in Japanese] J Integrated Med. 2013; 23 (12):1014-1017.
2. Seyahi E, Melikoglu M, Akman C, et al. Pulmonary artery involvement and associated lung disease in Behçet disease: a series of 47 patients. Medicine (Baltimore). 2012;91(1):35-48. PubMed
3. Olopade CO, Sekosan M, Schraufnagel DE. Giant cell arteritis manifesting as chronic cough and fever of unknown origin. Mayo Clin Proc. 1997;72(11):1048-1050. PubMed
4. Hellmann DB. Temporal arteritis: a cough, toothache, and tongue infarction. JAMA. 2002;287(22):2996-3000. PubMed
5. Karagiannis A, Mathiopoulou L, Tziomalos K, et al. Dry cough as first manifestation of giant-cell arteritis. J Am Geriatr Soc. 2006;54(12):1957-1958. PubMed
6. Aggarwal R, Ringold S, Khanna D, et al. Distinctions between diagnostic and classification criteria? Arthritis Care Res (Hoboken). 2015;67(7):891-897. PubMed
7. Rao JK, Allen NB, Pincus T. Limitations of the 1990 American College of Rheumatology classification criteria in the diagnosis of vasculitis. Ann Intern Med. 1998;129(5):345-352. PubMed
8. Davatchi F, Sadeghi Abdollahi B, Shahram F, Chams-Davatchi C, Shams H, Nadji A. Classification and Diagnosis Criteria for Behçet’s Disease. In: Emmi L, ed. Behçet’s Syndrome. From Pathogenesis to Treatment. Milan, Italy: Springer; 2014:189-198.
9. Criteria for diagnosis of Behcet’s disease. International Study Group for Behçet’s Disease. Lancet. 1990;335(8697):1078-1080. PubMed
10. Davatchi F, Assaad-Khalil S, Calamia KT, et al. The International Criteria for Behçet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338–347. PubMed
11. Suzuki Kurokawa M, Suzuki N. Behçet’s disease. Clin Exp Med. 2004;4(1):10-20. PubMed
© 2018 Society of Hospital Medicine