User login
In 2007, Falagas et al1 provided a systematic review of studies focusing on infection-related morbidity and mortality in patients with connective tissue diseases. Many of the studies reviewed were published prior to the introduction of biologic agents for the treatment of rheumatologic disorders. In 39 studies focusing on infection incidence, patient outcomes, or both in patients with systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), polymyositis/dermatomyositis, granulomatosis with polyangiitis (GPA, [Wegener’s granulomatosis]), and systemic sclerosis, serious infection developed in 29% of patients and 24% of these died due to the infection with a median attributable mortality of 5.2%. Most of the reported infections were common bacterial syndromes such as pneumonia or bacteremia, and opportunistic fungal (Pneumocystis) infections.
Similarly, in 2006 Alarcón2 reported that 25% to 50% of patients with SLE had significant morbidity primarily from common bacterial infections, with viral, fungal, and parasitic infection less common. Staphylococcus aureus was a common cause of soft tissue infection, septic arthritis, and bacteremia. Streptococcus pneumoniae typically caused respiratory infections, although meningitis and sepsis were reported with SLE. Gram-negative bacteria such as Escherichia coli, Klebsiella species, and Pseudomonas species usually caused urinary tract infections and nosocomial pneumonia. Other bacterial infections included Nocardia species, Mycobacterium tuberculosis, and, rarely, Listeria monocytogenes. The most common viral infection was herpes zoster. Fungal infections included Pneumocystis jirovecii (formerly known as Pneumocystis carinii) and Candida species.
In scleroderma, another connective tissue disease evaluated in the literature by Alarcón,2 reports of bacterial, viral, and fungal infections are limited to case reports. In scleroderma patients, viral infections with cytomegalovirus (CMV), parvovirus B19, and P jirovecii were similar to pathogens observed with SLE.
In polymyositis/dermatomyositis, gram-positive pneumonia affected 15% to 20% of patients and S aureus occurred frequently in the juvenile form of the disease. Herpes zoster was commonly observed, but CMV was relatively rare. Other viral infections included Coxsackie virus, parvovirus B19, and hepatitis C in polymyositis/dermatomyositis. Infection with P jirovecii is frequently fatal in these patients. Other fungal infections seen in polymyositis/dermatomyositis include candidiasis and histoplasmosis.2
Since the approval of antitumor necrosis factor (anti-TNF) agents for RA in the late 1990s, as well as other more recent biologic agents, there has been heightened awareness of infectious complications in rheumatologic patients. A major concern with the anti-TNF agents is the risk of granulomatous infection, particularly mycobacterial disease and dimorphic fungal infections such as histoplasmosis and coccidioidomycosis. Formation of granulomas is the major host defense against mycobacterial infection and is mediated in large part by TNF-alpha. The precise risk of infection associated with each of the various biologic agents is still under study, and rates from randomized trials have differed from postmarketing surveillance studies. Important pathogens associated with biologic agents include Nocardia, CMV, Listeria, Aspergillus, and JC virus (JCV).3,4 Delays in the diagnosis of these infections in immunocompromised patients have led to poor outcomes.
KEY PATHOGENS IN INFECTIONS OF IMMUNOCOMPROMISED HOSTS
Pneumocystis jirovecii
For many decades, P jirovecii was classified as a protozoan but, based on gene sequencing, the organism has been reclassified as a fungus. P jirovecii is a low-virulence, unicellular organism that is the causative agent of Pneumocystis pneumonia (PCP). Epidemiologically, primary infection most likely occurs in infants and children. Colonization may be transient, entering the airways and then resolving over a period of weeks or months. Alternatively, the organism may enter a latent state similar to tuberculosis with reactivation occurring during times of intense immunosuppression. However, molecular epidemiology studies show that new cases of PCP are likely environmentally acquired through multiple exposures rather than reactivation of latent infection.5,6 Transmission is thought to be airborne from person to person. Pathogenically, the trophic form of the organism attaches to type 1 alveolar cells and remains in the extracellular compartment of the alveoli. This colonization evokes an influx of inflammatory cells (CD8 cells, neutrophils, and macrophages). However, not all colonizations result in pneumonia—even in advanced human immunodeficiency virus (HIV) infection. While there is an innate immunity through alveolar macrophages and pulmonary surfactant, alveolar macrophage response is impaired in HIV when the CD4 count is low. Cell-mediated immunity is the main defense against progression to pneumonia with assistance from costimulatory molecules (such as CD28 and CD2) as well as B cells.
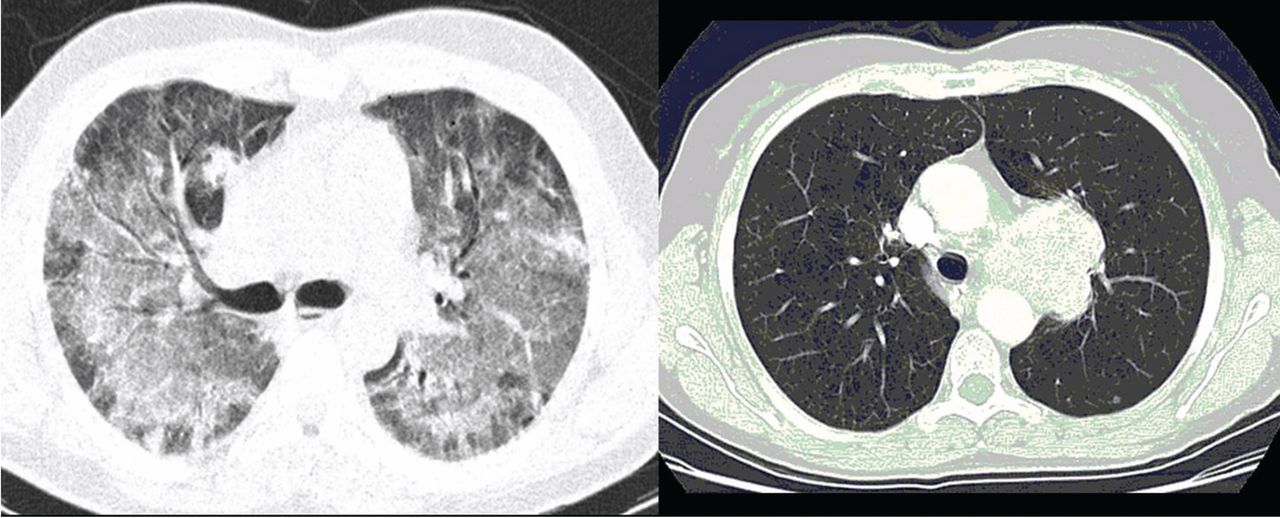
Laboratory diagnosis. P jirovecii cannot be grown in culture for clinical purposes, and it is extremely difficult to culture even in the research setting. Cytologic stains such as the Wright-Giemsa and methamine silver stains are the mainstay of laboratory diagnosis. The yield for P jirovecii from routine expectorated sputum is very low and some laboratories discourage this approach. The sensitivity of nebulized sputum using hypertonic saline ranges from 50% to 90%.9
In patients with acquired immune deficiency syndrome (AIDS), bronchoscopy provides 90% to 98% sensitivity by BAL. Transbronchial biopsy may provide some additional yield over BAL in a few situations, such as patients who have been receiving partial P jirovecii prophylaxis. Immunofluorescence techniques using monoclonal antibodies to P jirovecii are commercially available and are first-line diagnostic tools in some laboratories. Recently, polymerase chain reaction (PCR) assay has been introduced into clinical practice as a reproducible test with high sensitivity.
Primary therapy. Primary therapy for PCP consists of trimethoprim-sulfamethoxazole (TMP-SMX) or pentamidine. TMP-SMX is considered the drug of choice and is usually administered intravenously for 21 days in HIV patients and 14 days for non-HIV patients. The oral form may be used in patients with less severe PCP with a functioning gastrointestinal tract. Common adverse reactions to TMP-SMX include rash, Stevens-Johnson syndrome, neutropenia, changes in pulmonary function, and nausea/vomiting/diarrhea.10 Pentamidine is as effective as TMP-SMX, but is associated with renal toxicity, hypotension, severe hypoglycemia, cardiac arrhythmias, and diabetes.11 It is generally reserved for severe cases of PCP in patients who are allergic to or otherwise intolerant of sulfa. Other treatments include atovaquone and trimethoprim-dapsone. Adjunctive corticosteroids have been shown to be beneficial in moderate to severe PCP in HIV patients to reduce the local host inflammatory response to dead or dying organisms. Recent guidelines have recommended corticosteroids for HIV patients with PCP who have an arterial oxygen pressure of 70 mm Hg or less on room air, or an alveolar-arterial (A-a) gradient of oxygen 35 mm Hg or greater.12 Little is known about the role of adjunctive corticosteroids in non-HIV patients, given a lack of clinical studies.
Prevention. Recent estimates of disease burden from a meta-analysis of 11,900 patients with connective tissue diseases found PCP in 12% of patients with GPA, in 6% of those with polydermatomyositis, in 5% of those with SLE, and in 1% of those with RA.1 Mortality due to PCP is higher in patients with rheumatic diseases, ranging from 30% in RA to 63% in GPA, than in those with HIV (10% to 20%).13 One key risk factor predisposing patients with connective tissue diseases to infection with P jirovecii is recent corticosteroid use. Among patients with connective tissue disease, more than 90% of those infected with P jirovecii have recently received steroid therapy.14 Additionally, in almost all patients with P jirovecii, lymphopenia with absolute lymphocyte counts less than 1,000/mm3 is present.15
In patients with HIV, prophylaxis is initiated at a CD4 level of 200/mm3.13 However, the cutoff is less clear for non-HIV rheumatic patients. A cutoff of less than 300 cells/mm3 has been proposed for prophylaxis of PCP. However, at that range, approximately 50% of patients with connective tissue disease would remain above the threshold.13 One possible solution is to screen by PCR and treat colonization. Other algorithms have been proposed, but there is no general consensus on treatment of non-HIV rheumatic patients.13,16 Generally, prophylaxis should be considered in patients at the highest risk for PCP. These include patients taking prednisone at doses greater than 20 mg/day for 1 month plus a cytotoxic agent, a TNF inhibitor plus glucocorticoids, and methotrexate plus glucocorticoids in GPA.13
Nocardia asteroides and Nocardia species
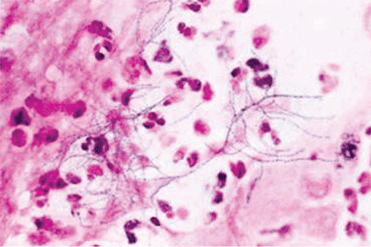
Classically, Nocardia infection results in abscess formation with infiltrates of polymorphonuclear cells, debris, and thin-walled abscesses. The most frequent site of primary infection is pulmonary. Characteristically, multiple pulmonary nodules or cavities are seen, and Nocardia should be considered in the differential diagnosis of an immunocompromised patient with nodular pneumonia. The nodules can also be masslike in appearance (greater than 2 cm). The presentation of new cavitary lung opacities with systemic symptoms may be mistaken for GPA.22Nocardia may disseminate to the central nervous system (CNS), skin, joints, and spine, usually causing suppurative infection at these sites. Nocardia has a very strong tropism for neural tissue. In the CNS, Nocardia can cause single or multiple brain abscesses that may be asymptomatic; patients with pulmonary nocardiosis require imaging to rule out occult CNS involvement.
Nocardia species are resistant to several antibiotics. The treatment of choice for Nocardia species is TMPSMX, but imipenem, amikacin, third-generation cephalosporins, and other options such as minocycline and linezolid may be considered depending on the species and the antimicrobial susceptibility pattern.
Histoplasma capsulatum
Histoplasma capsulatum is a dimorphic fungus that causes disease in both healthy and immunocompromised hosts. The organism differs from other pathogenic fungi in that it is an intracellular organism, mainly involving the reticuloendothelial system, and is rarely in the extracellular space. In the United States, infections are clustered endemically in areas such as the Mississippi and Ohio River Valleys, but infections are common worldwide. The fungus is found in soil, mulch, bird excrement, and bat guano. Asymptomatic or mild infections are common in healthy persons residing in endemic areas and occur on a sporadic basis. Epidemics can occur when contaminated material is aerosolized. Histoplasmosis is also an opportunistic infection in patients with impaired T-cell immunity such as persons with AIDS, organ transplant recipients, hematologic malignancies, and corticosteroid use. Clinically significant cases of histoplasmosis have been described in patients with RA while receiving methotrexate alone, corticosteroids alone, and combinations of disease-modifying agents.23 Histoplasmosis was recently identified in 240 patients in association with TNF inhibitors, translating to 17 per 100,000 patients treated with infliximab.21,24
Pathogenesis. Infection initially occurs through inhalation of contaminated material from the environment, primarily causing pulmonary infection. The organism converts from a mold form in the environment to a pathogenic yeast form in the host. Once inhaled, the mediastinal lymph nodes provide the first line of defense. Following draining of the lymph nodes, the organism enters the bloodstream in both immunocompetent and immunosuppressed patients. It is spread hematogenously into the spleen, liver, and reticulo-endothelial system, where it is eventually cleared. In immunocompetent patients, cellular immunity limits infection within 7 to 14 days and humoral immunity is not protective.25 Granuloma formation is the hallmark of host defense.
Spectrum of illness. Histoplasmosis is associated with a wide spectrum of illness, with presentation ranging from asymptomatic to mild pulmonary illness to overwhelming pneumonia. Symptomatic pulmonary histoplasmosis typically presents with fever, flulike symptoms, and cough, often with retrosternal chest pain. X-rays show patchy or nodular infiltrates, with hilar or mediastinal lymphadenopathy. In some cases the lung parenchyma is clear and the main feature is fever and bilateral hilar adenopathy. Pulmonary histoplasmosis may be difficult to distinguish from sarcoidosis and tuberculosis. Extrapulmonary disease can present as hepatitis, infective endocarditis, and chronic meningitis. In immunocompromised patients, histoplasmosis can present as a progressive disseminated disease which can be acute, subacute, or chronic. Chronic disseminated histoplasmosis is characterized by cough, persistent fever, wasting, hepatosplenomegaly, oral ulcerations, and progressive cytopenias. Acute disseminated histoplasmosis has a much more fulminant course characterized by respiratory insufficiency, hypotension, multisystem organ failure, coagulopathies, and encephalopathy. Histoplasmosis is primarily a pulmonary disease, but in disseminated disease more than 50% of patients have no pulmonary symptoms and 30% may have normal chest x-rays.26 In one series of infliximab-related cases (n = 10), all came from an endemic area 1 week to 6 months after the first dose of infliximab. Patients presented with cough, fever, and shortness of breath.27 The pathogenesis of histoplasmosis in patients receiving TNF inhibitors is not entirely clear; such patients may be suffering a new primary infection, a reinfection, or, least likely, reactivation of latent infection.
Definitive diagnosis requires culture confirmation from appropriate body fluids or identification of characteristic yeast forms from histopathologic sections of tissue biopsies. Serologic tests may also be used to confirm the diagnosis. Detection of H and M precipitins or bands by immunodiffusion is a routine test in many laboratories. M bands are present in 50% of acute cases but their presence does not distinguish acute from remote infection. H bands are present in only 10% of all acute cases, but their presence is very specific for acute histoplasmosis.28
When looking at complement fixation antibodies to yeast (HY) and mycelial (HMy) forms in pulmonary histoplasmosis, a fourfold rise in titer establishes the diagnosis retrospectively, and a single titer greater than 1:32 is strongly suggestive of active infection. However, in progressive disseminated histoplasmosis, the complement fixation antibodies are frequently negative.29 Detection of antigen in urine and serum by enzyme immunoassay has become a mainstay of diagnosis, with a sensitivity of approximately 90% in progressive disseminated disease.30 Of note, most cases of histoplasmosis associated with biologic agents have detectable urinary antigen tests.
Treatment. Acute pulmonary histoplasmosis is usually self-limited, requiring no treatment. The 2007 Infective Diseases Society of America (IDSA) guidelines recommend observation alone in most cases of mild to moderate pulmonary histoplasmosis unless symptoms persist longer than 1 month. For moderately severe or severe acute pulmonary histoplasmosis, the IDSA recommends lipid formulations of amphotericin B (3.0 to 5.0 mg/kg/day) or deosycholate amphotericin B (0.7 to 1.0 mg/kg/day) for 1 to 2 weeks followed by itraconazole 200 mg twice daily for a total of 12 weeks. Methylprednisolone at a dose of 0.5 to 1.0 mg/kg/day intravenously for 1 to 2 weeks is also recommended. For moderately severe to severe disseminated histoplasmosis, the IDSA recommends lipid formulations of amphotericin B (3.0 mg/kg/day) for 1 to 2 weeks followed by oral itraconazole 200 mg three times daily for 3 days and then 200 mg twice daily for a total of at least 12 months.31 Commonly, the immunosuppressive agent is held during treatment.
Aspergillus species
Another emerging pathogen is Aspergillus species—a ubiquitous mold spread by aerosols of spores. There are many different species of Aspergillus, but the most common human pathogens include A fumigates, A niger, and A flavus. To date, 39 cases of Aspergillus infection associated with infliximab and etanercept have been reported in the Adverse Event Reporting System, translating to 9 to 12 cases per 100,000 patients.21
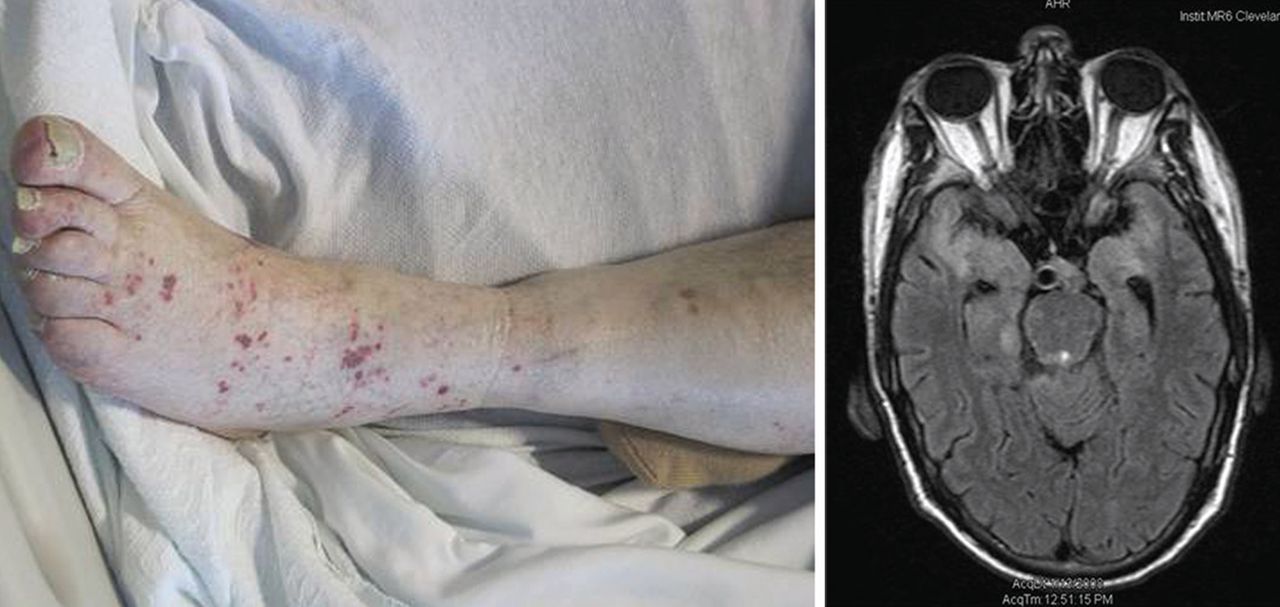
Varicella zoster
JC virus
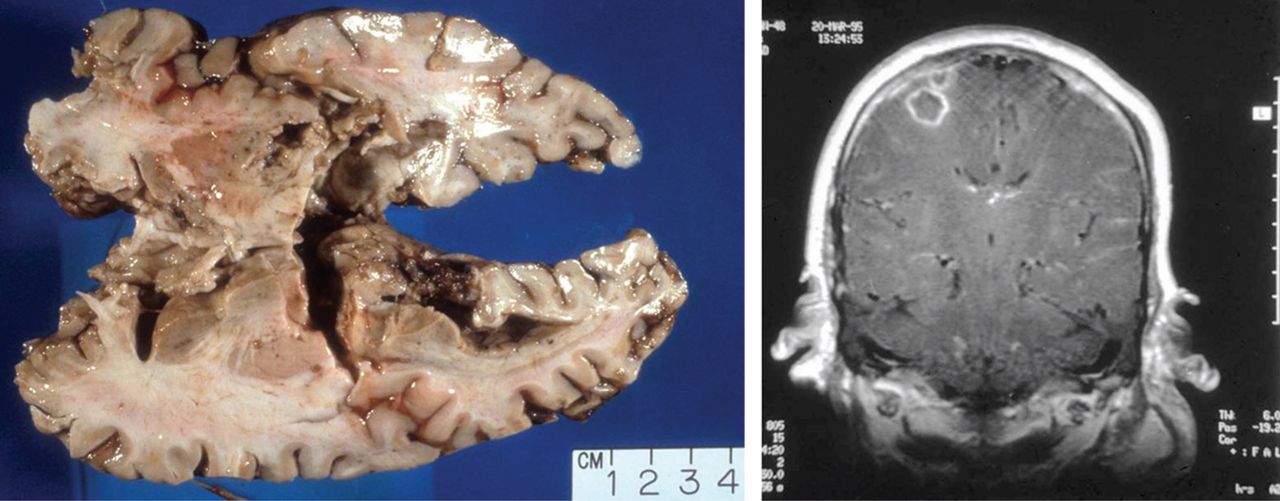
More than 80% of adults are seropositive for JCV, a DNA virus of the genus Polyomavirus that causes lytic infection of oligodendrocytes.34 In immunocompromised hosts, JCV causes progressive multifocal leukoencephalopathy (PML), a rare but devastating demyelinating disease. PML was first described in malignancy, leukemia, and various other immunocompromised states, prior to its strong association with AIDS in the 1980s. More recently, JCV has been associated with natalizumab for multiple sclerosis and Crohn disease, rituximab for oncology patients, efalizumab for psoriasis,35 and mycophenolate mofetil for transplant recipients.36
In 2006 the US Food and Drug Administration issued a safety alert regarding PML in two patients with SLE treated with rituximab and other immunosuppressives.37 In a review of PML in rheumatic disease, 36 cases were identified in patients who had not previously received a biologic agent. Most of these patients (60%) had SLE.38 Of these, many had little or no immunosuppression over the 6 months prior to the diagnosis of PML, suggesting that SLE itself may predispose to PML. Interestingly, PML is rarely associated with TNF inhibitors.
Classic presentation of PML includes motor weakness, aphasia, dysarthria, vision loss, and cognitive loss. Atypical presentation includes seizures, headaches, and brainstem involvement. PML usually spares the optic nerves, spinal cord, peripheral nerves, and muscles. In persons with underlying rheumatic diseases, PML can be difficult to distinguish from neuropsychiatric SLE or CNS vasculitis.
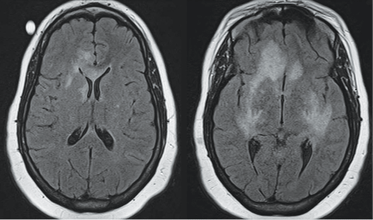
Treatment. In clinical trials no antiviral agent has been effective in the treatment of PML. In HIV patients who develop PML, highly active antiretroviral therapy should be initiated (if antiretroviral-naïve) or existing antiviral regimens optimized. Antiretroviral therapy in this situation may stabilize disease and possibly increase survival.42 For HIV-negative patients who develop PML, the cornerstone of management is immediate decrease or discontinuation of immunosuppression.43 Several adjunctive measures have been reported mainly in natalizumab-associated PML, including corticosteroids, mirtazapine, plasma exchange, and others.
VACCINES
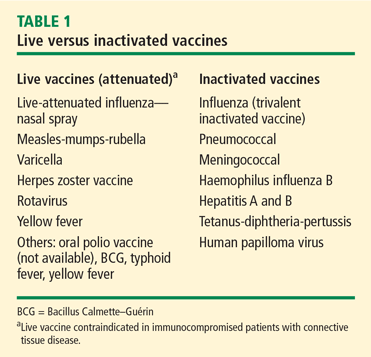
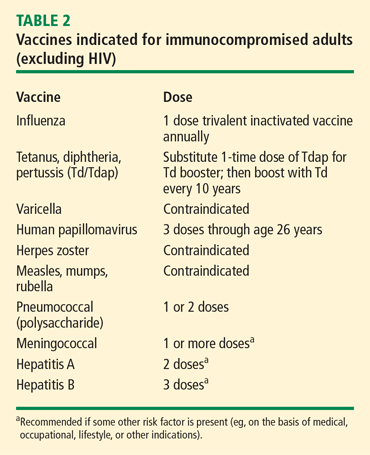
Vaccination is important in the prevention of infectious disease in immunocompromised patients with connective tissue diseases. Because live vaccines are contraindicated in immunocompromised patients, inactivated or component vaccines should be used. It is recommended that patients who will start immunosuppressive therapy be vaccinated 2 to 4 weeks before beginning therapy. If this is not possible, vaccination should be administered during disease remission, 3 months after immunosuppression and 1 to 3 months after administration of high-dose corticosteroids.
- Short-term (less than 14 days)
- At a dose of less than 20 mg/day of prednisone or equivalent
- Long-term on alternate days with short-acting preparations
- At a physiologic dose of prednisone
- Topical, inhaled, intra-articular, bursal, or via tendon.44
Until definitive guidelines are developed, practitioners must evaluate and treat each patient individually to maximize the efficacy of disease treatments while preventing infection morbidity and mortality in their patients with connective tissue diseases.
- Falagas ME, Manta KG, Betsi GI, Pappas G. Infection-related morbidity and mortality in patients with connective tissue diseases: a systematic review. Clin Rheumatol 2007; 26:663–670.
- Alarcón GS. Infections in systemic connective tissue diseases: systemic lupus erythematosus, scleroderma, and polymyositis/dermatomyositis. Infect Dis Clin North Am 2006; 20:849–875.
- Crum NF, Lederman ER, Wallace MR. Infections associated with tumor necrosis factor-alpha antagonists. Medicine (Baltimore) 2005; 84:291–302.
- Rychly DJ, DiPiro JT. Infections associated with tumor necrosis factor-alpha antagonists. Pharmacotherapy 2005; 25:1181–1192.
- Wakefield AE, Lindley AR, Ambrose HE, Denis CM, Miller RF. Limited asymptomatic carriage of Pneumocystis jiroveci in human immunodeficiency virus-infected patients [published online ahead of print March 6, 2003]. J Infect Dis 2003; 187:901–908. doi: 10.1086/368165
- Beard CB, Carter JL, Keely SP, et al. Genetic variation in Pneumocystis carinii isolates from different geographic regions: implications for transmission. Emerg Infect Dis 2000; 6:265–272.
- Walzer PD, Smulian AG. Pneumocystis species. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, Pa: Elsevier Churchill Livingstone; 2009.
- Hartman TE, Primack SL, Müller NL, Staples CA. Diagnosis of thoracic complications in AIDS: accuracy of CT. Am J Roentgenol 1994; 162:547–553.
- Shelhamer JH, Gill VJ, Quinn TC, et al. The laboratory evaluation of opportunistic pulmonary infections. Ann Intern Med 1996; 124:585–599.
- Wharton JM, Coleman DL, Wofsy CB, et al. Trimethoprim-sulfamethoxazole or pentamidine for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. Ann Intern Med 1986; 105:37–44.
- Stein DS, Stevens RC. Treatment-associated toxicities: incidence and mechanisms. In:Sattler FR, Walzer PD, eds. Pneumocystis carinii. London: Bailliere Tindall; 1995:505–530.
- Consensus statement on the use of corticosteroids as adjunctive therapy for Pneumocystis pneumonia in the acquired immunodeficiency syndrome. The National Institutes of Health-University of California Expert Panel for Corticosteroids as Adjunctive Therapy for Pneumocystis Pneumonia. N Engl J Med 1990; 323:1500–1504.
- Stamp LK, Hurst M. Is there a role for consensus guidelines for P. jiroveci pneumonia prophylaxis in immunosuppressed patients with rheumatic diseases? J Rheumatol 2010; 37:686–688.
- Yale S, Limper A. Pneumocystis carinii pneumonia in patients without acquired immunodeficiency syndrome: associated illnesses and prior corticosteroid therapy. Mayo Clin Proc 1996; 71:5–13.
- Sowden E, Carmichael A. Autoimmune inflammatory disorders, systemic corticosteroids and Pneumocystis pneumonia: a strategy for prevention [published online October 16, 2004]. BMC Infect Dis 2004; 4:42. doi: 10.1186/1471-2334-4-42
- Cettomai D, Gelber AC, Christopher-Stine L. A survey of rheumatologists’ practice for prescribing Pneumocystis prophylaxis. J Rheumatol 2010; 37:792–799.
- Keegan JM, Byrd JW. Nocardiosis associated with low dose methotrexate for rheumatoid arthritis. J Rheumatol 1988; 15:1585–1586.
- Gruberg L, Thaler M, Rozenman J, et al. Nocardia asteroides infection complicating rheumatoid arthritis. J Rheumatol 1991; 18:459–461.
- Corneliessen JJ, Bakker LJ, van der Veen MJ, et al. Nocardia asteroides pneumonia complicating low dose methotrexate treatment of refractory rheumatoid arthritis. Ann Rheum Dis 1991; 50;642–644.
- Silva C, Faccioli LH. Tumor necrosis factor and macrophage activation are important in clearance of Nocardia brasiliensis from the livers and spleens of mice. Infect Immun 1992; 60:3566–3570.
- Wallis RS, Broder MS, Wong JY, Hanson ME, Beenhouwer DO. Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin Infect Dis 2004; 38:1261–1265.
- Gibb W, Williams A. Nocardiosis mimicking Wegener’s granulomatosis. Scand J Infect Dis 1986; 18:583–585.
- Olson TC, Bongartz T, Crowson CS, Roberts GD, Orenstein R, Matteson EI. Histoplasmosis infection in patients with rheumatoid arthritis, 1998–2009 [published online May 23, 2011]. BMC Infectious Diseases 2011; 11:145. doi: 10.1186/1471-2334-11-145
- Information for Healthcare Professionals: Cimzia (certolizumab pegol), Enbrel etanercept), Humira (adalimumab), and Remicade (infliximab). U.S. Food and Drug Administration Web site. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124185.htm. Updated January 25, 2010. Accessed September 27, 2012.
- Paya CV, Roberts GD, Cockerill FR. Transient fungemia in acute pulmonary histoplasmosis: detection by new blood-culturing techniques. J Infect Dis 1987; 156:313–315.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Lee JH, Slifman NR, Gershon SK, et al. Life-threatening histoplasmosis complicating immunotherapy with tumor necrosis factor alpha antagonists infliximab and etanercept. Arthritis Rheum 2002; 46:2565–2570.
- Picardi JL, Kauffman CA, Schwarz J, Phair JP. Detection of precipitating antibodies to Histoplasma capsulatum by counterimmunoelectrophoresis. Am Rev Respir Dis 1976; 114:171–176.
- Deepe GS. Histoplasma capsulatum. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, Pa: Elsevier Churchill Livingstone; 2009.
- Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev 2007; 20:115–132.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America [published online ahead of print August 27, 2007]. Clin Infect Dis 2007; 45:807–825. doi: 10.1086/521259
- Johnson RW. Herpes zoster and postherpetic neuralgia. Expert Rev Vaccines 2010; 9( 3 suppl):21–26.
- Wolfe F, Michaud K, Chakravarty EF. Rates and predictors of herpes zoster in patients with rheumatoid arthritis and non-inflammatory musculoskeletal disorders. Rheumatology 2006; 45:1370–1375.
- Weber T, Trebst C, Frye S, et al. Analysis of the systemic and intrathecal humoral immune response in progressive multifocal leukoencephalopathy. J Infect Dis 1997; 176:250–254.
- Kothary N, Diak IL, Brinker A, Bezabeh S, Avigan M, Dal Pan G. Progressive multifocal leukoencephalopathy associated with efalizumab use in psoriasis patients. J Am Acad Dermatol 2011; 65:546–551.
- Neff RT, Hurst FP, Falta EM, et al. Progressive multifocal leukoencephalopathy and use of mycophenolate mofetil after kidney transplantation. Transplantation 2008; 86:1474–1478.
- Rituxan warning. FDA Consum 2007; 41:3.
- Calabrese LH, Molloy ES, Huang D, Ransohoff RM. Progressive multifocal leukoencephalopathy in rheumatic diseases. Arthritis Rheum 2007; 56:2116–2128.
- Cinque P, Scarpellini P, Vago L, Linde A, Lazzarin A. Diagnosis of central nervous system complications in HIV-infected patients: cerebrospinal fluid analysis by the polymerase chain reaction. AIDS 1997; 11:1–17.
- Ryschkewitsch C, Jensen P, Hou J, Fahle G, Fischer S, Major EO. Comparison of PCR-southern hybridization and quantitative real-time PCR for the detection of JC and BK viral nucleotide sequences in urine and cerebrospinal fluid. J Virol Methods 2004; 121:217–221.
- Major EO. History and current concepts in the pathogenesis of PML. Cleve Clin J Med 2011; 78( suppl 2):S3–S7.
- Antinori A, Ammassari A, Giancola ML, et al. Epidemiology and prognosis of AIDS-associated progressive multifocal leukoencephalopathy in the HAART era. J Neurovirol 2001; 7:323–328.
- Calabrese L. A rational approach to PML for the clinician. Cleve Clin J Med 2011; 78 (suppl 2):S38–S41.
- Kroger AT, Sumaya CV, Pickering LK, Atkinson WL. General recommendations on immunization. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2011; 60:1–60.
In 2007, Falagas et al1 provided a systematic review of studies focusing on infection-related morbidity and mortality in patients with connective tissue diseases. Many of the studies reviewed were published prior to the introduction of biologic agents for the treatment of rheumatologic disorders. In 39 studies focusing on infection incidence, patient outcomes, or both in patients with systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), polymyositis/dermatomyositis, granulomatosis with polyangiitis (GPA, [Wegener’s granulomatosis]), and systemic sclerosis, serious infection developed in 29% of patients and 24% of these died due to the infection with a median attributable mortality of 5.2%. Most of the reported infections were common bacterial syndromes such as pneumonia or bacteremia, and opportunistic fungal (Pneumocystis) infections.
Similarly, in 2006 Alarcón2 reported that 25% to 50% of patients with SLE had significant morbidity primarily from common bacterial infections, with viral, fungal, and parasitic infection less common. Staphylococcus aureus was a common cause of soft tissue infection, septic arthritis, and bacteremia. Streptococcus pneumoniae typically caused respiratory infections, although meningitis and sepsis were reported with SLE. Gram-negative bacteria such as Escherichia coli, Klebsiella species, and Pseudomonas species usually caused urinary tract infections and nosocomial pneumonia. Other bacterial infections included Nocardia species, Mycobacterium tuberculosis, and, rarely, Listeria monocytogenes. The most common viral infection was herpes zoster. Fungal infections included Pneumocystis jirovecii (formerly known as Pneumocystis carinii) and Candida species.
In scleroderma, another connective tissue disease evaluated in the literature by Alarcón,2 reports of bacterial, viral, and fungal infections are limited to case reports. In scleroderma patients, viral infections with cytomegalovirus (CMV), parvovirus B19, and P jirovecii were similar to pathogens observed with SLE.
In polymyositis/dermatomyositis, gram-positive pneumonia affected 15% to 20% of patients and S aureus occurred frequently in the juvenile form of the disease. Herpes zoster was commonly observed, but CMV was relatively rare. Other viral infections included Coxsackie virus, parvovirus B19, and hepatitis C in polymyositis/dermatomyositis. Infection with P jirovecii is frequently fatal in these patients. Other fungal infections seen in polymyositis/dermatomyositis include candidiasis and histoplasmosis.2
Since the approval of antitumor necrosis factor (anti-TNF) agents for RA in the late 1990s, as well as other more recent biologic agents, there has been heightened awareness of infectious complications in rheumatologic patients. A major concern with the anti-TNF agents is the risk of granulomatous infection, particularly mycobacterial disease and dimorphic fungal infections such as histoplasmosis and coccidioidomycosis. Formation of granulomas is the major host defense against mycobacterial infection and is mediated in large part by TNF-alpha. The precise risk of infection associated with each of the various biologic agents is still under study, and rates from randomized trials have differed from postmarketing surveillance studies. Important pathogens associated with biologic agents include Nocardia, CMV, Listeria, Aspergillus, and JC virus (JCV).3,4 Delays in the diagnosis of these infections in immunocompromised patients have led to poor outcomes.
KEY PATHOGENS IN INFECTIONS OF IMMUNOCOMPROMISED HOSTS
Pneumocystis jirovecii
For many decades, P jirovecii was classified as a protozoan but, based on gene sequencing, the organism has been reclassified as a fungus. P jirovecii is a low-virulence, unicellular organism that is the causative agent of Pneumocystis pneumonia (PCP). Epidemiologically, primary infection most likely occurs in infants and children. Colonization may be transient, entering the airways and then resolving over a period of weeks or months. Alternatively, the organism may enter a latent state similar to tuberculosis with reactivation occurring during times of intense immunosuppression. However, molecular epidemiology studies show that new cases of PCP are likely environmentally acquired through multiple exposures rather than reactivation of latent infection.5,6 Transmission is thought to be airborne from person to person. Pathogenically, the trophic form of the organism attaches to type 1 alveolar cells and remains in the extracellular compartment of the alveoli. This colonization evokes an influx of inflammatory cells (CD8 cells, neutrophils, and macrophages). However, not all colonizations result in pneumonia—even in advanced human immunodeficiency virus (HIV) infection. While there is an innate immunity through alveolar macrophages and pulmonary surfactant, alveolar macrophage response is impaired in HIV when the CD4 count is low. Cell-mediated immunity is the main defense against progression to pneumonia with assistance from costimulatory molecules (such as CD28 and CD2) as well as B cells.
Laboratory diagnosis. P jirovecii cannot be grown in culture for clinical purposes, and it is extremely difficult to culture even in the research setting. Cytologic stains such as the Wright-Giemsa and methamine silver stains are the mainstay of laboratory diagnosis. The yield for P jirovecii from routine expectorated sputum is very low and some laboratories discourage this approach. The sensitivity of nebulized sputum using hypertonic saline ranges from 50% to 90%.9
In patients with acquired immune deficiency syndrome (AIDS), bronchoscopy provides 90% to 98% sensitivity by BAL. Transbronchial biopsy may provide some additional yield over BAL in a few situations, such as patients who have been receiving partial P jirovecii prophylaxis. Immunofluorescence techniques using monoclonal antibodies to P jirovecii are commercially available and are first-line diagnostic tools in some laboratories. Recently, polymerase chain reaction (PCR) assay has been introduced into clinical practice as a reproducible test with high sensitivity.
Primary therapy. Primary therapy for PCP consists of trimethoprim-sulfamethoxazole (TMP-SMX) or pentamidine. TMP-SMX is considered the drug of choice and is usually administered intravenously for 21 days in HIV patients and 14 days for non-HIV patients. The oral form may be used in patients with less severe PCP with a functioning gastrointestinal tract. Common adverse reactions to TMP-SMX include rash, Stevens-Johnson syndrome, neutropenia, changes in pulmonary function, and nausea/vomiting/diarrhea.10 Pentamidine is as effective as TMP-SMX, but is associated with renal toxicity, hypotension, severe hypoglycemia, cardiac arrhythmias, and diabetes.11 It is generally reserved for severe cases of PCP in patients who are allergic to or otherwise intolerant of sulfa. Other treatments include atovaquone and trimethoprim-dapsone. Adjunctive corticosteroids have been shown to be beneficial in moderate to severe PCP in HIV patients to reduce the local host inflammatory response to dead or dying organisms. Recent guidelines have recommended corticosteroids for HIV patients with PCP who have an arterial oxygen pressure of 70 mm Hg or less on room air, or an alveolar-arterial (A-a) gradient of oxygen 35 mm Hg or greater.12 Little is known about the role of adjunctive corticosteroids in non-HIV patients, given a lack of clinical studies.
Prevention. Recent estimates of disease burden from a meta-analysis of 11,900 patients with connective tissue diseases found PCP in 12% of patients with GPA, in 6% of those with polydermatomyositis, in 5% of those with SLE, and in 1% of those with RA.1 Mortality due to PCP is higher in patients with rheumatic diseases, ranging from 30% in RA to 63% in GPA, than in those with HIV (10% to 20%).13 One key risk factor predisposing patients with connective tissue diseases to infection with P jirovecii is recent corticosteroid use. Among patients with connective tissue disease, more than 90% of those infected with P jirovecii have recently received steroid therapy.14 Additionally, in almost all patients with P jirovecii, lymphopenia with absolute lymphocyte counts less than 1,000/mm3 is present.15
In patients with HIV, prophylaxis is initiated at a CD4 level of 200/mm3.13 However, the cutoff is less clear for non-HIV rheumatic patients. A cutoff of less than 300 cells/mm3 has been proposed for prophylaxis of PCP. However, at that range, approximately 50% of patients with connective tissue disease would remain above the threshold.13 One possible solution is to screen by PCR and treat colonization. Other algorithms have been proposed, but there is no general consensus on treatment of non-HIV rheumatic patients.13,16 Generally, prophylaxis should be considered in patients at the highest risk for PCP. These include patients taking prednisone at doses greater than 20 mg/day for 1 month plus a cytotoxic agent, a TNF inhibitor plus glucocorticoids, and methotrexate plus glucocorticoids in GPA.13
Nocardia asteroides and Nocardia species
Classically, Nocardia infection results in abscess formation with infiltrates of polymorphonuclear cells, debris, and thin-walled abscesses. The most frequent site of primary infection is pulmonary. Characteristically, multiple pulmonary nodules or cavities are seen, and Nocardia should be considered in the differential diagnosis of an immunocompromised patient with nodular pneumonia. The nodules can also be masslike in appearance (greater than 2 cm). The presentation of new cavitary lung opacities with systemic symptoms may be mistaken for GPA.22Nocardia may disseminate to the central nervous system (CNS), skin, joints, and spine, usually causing suppurative infection at these sites. Nocardia has a very strong tropism for neural tissue. In the CNS, Nocardia can cause single or multiple brain abscesses that may be asymptomatic; patients with pulmonary nocardiosis require imaging to rule out occult CNS involvement.
Nocardia species are resistant to several antibiotics. The treatment of choice for Nocardia species is TMPSMX, but imipenem, amikacin, third-generation cephalosporins, and other options such as minocycline and linezolid may be considered depending on the species and the antimicrobial susceptibility pattern.
Histoplasma capsulatum
Histoplasma capsulatum is a dimorphic fungus that causes disease in both healthy and immunocompromised hosts. The organism differs from other pathogenic fungi in that it is an intracellular organism, mainly involving the reticuloendothelial system, and is rarely in the extracellular space. In the United States, infections are clustered endemically in areas such as the Mississippi and Ohio River Valleys, but infections are common worldwide. The fungus is found in soil, mulch, bird excrement, and bat guano. Asymptomatic or mild infections are common in healthy persons residing in endemic areas and occur on a sporadic basis. Epidemics can occur when contaminated material is aerosolized. Histoplasmosis is also an opportunistic infection in patients with impaired T-cell immunity such as persons with AIDS, organ transplant recipients, hematologic malignancies, and corticosteroid use. Clinically significant cases of histoplasmosis have been described in patients with RA while receiving methotrexate alone, corticosteroids alone, and combinations of disease-modifying agents.23 Histoplasmosis was recently identified in 240 patients in association with TNF inhibitors, translating to 17 per 100,000 patients treated with infliximab.21,24
Pathogenesis. Infection initially occurs through inhalation of contaminated material from the environment, primarily causing pulmonary infection. The organism converts from a mold form in the environment to a pathogenic yeast form in the host. Once inhaled, the mediastinal lymph nodes provide the first line of defense. Following draining of the lymph nodes, the organism enters the bloodstream in both immunocompetent and immunosuppressed patients. It is spread hematogenously into the spleen, liver, and reticulo-endothelial system, where it is eventually cleared. In immunocompetent patients, cellular immunity limits infection within 7 to 14 days and humoral immunity is not protective.25 Granuloma formation is the hallmark of host defense.
Spectrum of illness. Histoplasmosis is associated with a wide spectrum of illness, with presentation ranging from asymptomatic to mild pulmonary illness to overwhelming pneumonia. Symptomatic pulmonary histoplasmosis typically presents with fever, flulike symptoms, and cough, often with retrosternal chest pain. X-rays show patchy or nodular infiltrates, with hilar or mediastinal lymphadenopathy. In some cases the lung parenchyma is clear and the main feature is fever and bilateral hilar adenopathy. Pulmonary histoplasmosis may be difficult to distinguish from sarcoidosis and tuberculosis. Extrapulmonary disease can present as hepatitis, infective endocarditis, and chronic meningitis. In immunocompromised patients, histoplasmosis can present as a progressive disseminated disease which can be acute, subacute, or chronic. Chronic disseminated histoplasmosis is characterized by cough, persistent fever, wasting, hepatosplenomegaly, oral ulcerations, and progressive cytopenias. Acute disseminated histoplasmosis has a much more fulminant course characterized by respiratory insufficiency, hypotension, multisystem organ failure, coagulopathies, and encephalopathy. Histoplasmosis is primarily a pulmonary disease, but in disseminated disease more than 50% of patients have no pulmonary symptoms and 30% may have normal chest x-rays.26 In one series of infliximab-related cases (n = 10), all came from an endemic area 1 week to 6 months after the first dose of infliximab. Patients presented with cough, fever, and shortness of breath.27 The pathogenesis of histoplasmosis in patients receiving TNF inhibitors is not entirely clear; such patients may be suffering a new primary infection, a reinfection, or, least likely, reactivation of latent infection.
Definitive diagnosis requires culture confirmation from appropriate body fluids or identification of characteristic yeast forms from histopathologic sections of tissue biopsies. Serologic tests may also be used to confirm the diagnosis. Detection of H and M precipitins or bands by immunodiffusion is a routine test in many laboratories. M bands are present in 50% of acute cases but their presence does not distinguish acute from remote infection. H bands are present in only 10% of all acute cases, but their presence is very specific for acute histoplasmosis.28
When looking at complement fixation antibodies to yeast (HY) and mycelial (HMy) forms in pulmonary histoplasmosis, a fourfold rise in titer establishes the diagnosis retrospectively, and a single titer greater than 1:32 is strongly suggestive of active infection. However, in progressive disseminated histoplasmosis, the complement fixation antibodies are frequently negative.29 Detection of antigen in urine and serum by enzyme immunoassay has become a mainstay of diagnosis, with a sensitivity of approximately 90% in progressive disseminated disease.30 Of note, most cases of histoplasmosis associated with biologic agents have detectable urinary antigen tests.
Treatment. Acute pulmonary histoplasmosis is usually self-limited, requiring no treatment. The 2007 Infective Diseases Society of America (IDSA) guidelines recommend observation alone in most cases of mild to moderate pulmonary histoplasmosis unless symptoms persist longer than 1 month. For moderately severe or severe acute pulmonary histoplasmosis, the IDSA recommends lipid formulations of amphotericin B (3.0 to 5.0 mg/kg/day) or deosycholate amphotericin B (0.7 to 1.0 mg/kg/day) for 1 to 2 weeks followed by itraconazole 200 mg twice daily for a total of 12 weeks. Methylprednisolone at a dose of 0.5 to 1.0 mg/kg/day intravenously for 1 to 2 weeks is also recommended. For moderately severe to severe disseminated histoplasmosis, the IDSA recommends lipid formulations of amphotericin B (3.0 mg/kg/day) for 1 to 2 weeks followed by oral itraconazole 200 mg three times daily for 3 days and then 200 mg twice daily for a total of at least 12 months.31 Commonly, the immunosuppressive agent is held during treatment.
Aspergillus species
Another emerging pathogen is Aspergillus species—a ubiquitous mold spread by aerosols of spores. There are many different species of Aspergillus, but the most common human pathogens include A fumigates, A niger, and A flavus. To date, 39 cases of Aspergillus infection associated with infliximab and etanercept have been reported in the Adverse Event Reporting System, translating to 9 to 12 cases per 100,000 patients.21
Varicella zoster
JC virus
More than 80% of adults are seropositive for JCV, a DNA virus of the genus Polyomavirus that causes lytic infection of oligodendrocytes.34 In immunocompromised hosts, JCV causes progressive multifocal leukoencephalopathy (PML), a rare but devastating demyelinating disease. PML was first described in malignancy, leukemia, and various other immunocompromised states, prior to its strong association with AIDS in the 1980s. More recently, JCV has been associated with natalizumab for multiple sclerosis and Crohn disease, rituximab for oncology patients, efalizumab for psoriasis,35 and mycophenolate mofetil for transplant recipients.36
In 2006 the US Food and Drug Administration issued a safety alert regarding PML in two patients with SLE treated with rituximab and other immunosuppressives.37 In a review of PML in rheumatic disease, 36 cases were identified in patients who had not previously received a biologic agent. Most of these patients (60%) had SLE.38 Of these, many had little or no immunosuppression over the 6 months prior to the diagnosis of PML, suggesting that SLE itself may predispose to PML. Interestingly, PML is rarely associated with TNF inhibitors.
Classic presentation of PML includes motor weakness, aphasia, dysarthria, vision loss, and cognitive loss. Atypical presentation includes seizures, headaches, and brainstem involvement. PML usually spares the optic nerves, spinal cord, peripheral nerves, and muscles. In persons with underlying rheumatic diseases, PML can be difficult to distinguish from neuropsychiatric SLE or CNS vasculitis.
Treatment. In clinical trials no antiviral agent has been effective in the treatment of PML. In HIV patients who develop PML, highly active antiretroviral therapy should be initiated (if antiretroviral-naïve) or existing antiviral regimens optimized. Antiretroviral therapy in this situation may stabilize disease and possibly increase survival.42 For HIV-negative patients who develop PML, the cornerstone of management is immediate decrease or discontinuation of immunosuppression.43 Several adjunctive measures have been reported mainly in natalizumab-associated PML, including corticosteroids, mirtazapine, plasma exchange, and others.
VACCINES
Vaccination is important in the prevention of infectious disease in immunocompromised patients with connective tissue diseases. Because live vaccines are contraindicated in immunocompromised patients, inactivated or component vaccines should be used. It is recommended that patients who will start immunosuppressive therapy be vaccinated 2 to 4 weeks before beginning therapy. If this is not possible, vaccination should be administered during disease remission, 3 months after immunosuppression and 1 to 3 months after administration of high-dose corticosteroids.
- Short-term (less than 14 days)
- At a dose of less than 20 mg/day of prednisone or equivalent
- Long-term on alternate days with short-acting preparations
- At a physiologic dose of prednisone
- Topical, inhaled, intra-articular, bursal, or via tendon.44
Until definitive guidelines are developed, practitioners must evaluate and treat each patient individually to maximize the efficacy of disease treatments while preventing infection morbidity and mortality in their patients with connective tissue diseases.
In 2007, Falagas et al1 provided a systematic review of studies focusing on infection-related morbidity and mortality in patients with connective tissue diseases. Many of the studies reviewed were published prior to the introduction of biologic agents for the treatment of rheumatologic disorders. In 39 studies focusing on infection incidence, patient outcomes, or both in patients with systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), polymyositis/dermatomyositis, granulomatosis with polyangiitis (GPA, [Wegener’s granulomatosis]), and systemic sclerosis, serious infection developed in 29% of patients and 24% of these died due to the infection with a median attributable mortality of 5.2%. Most of the reported infections were common bacterial syndromes such as pneumonia or bacteremia, and opportunistic fungal (Pneumocystis) infections.
Similarly, in 2006 Alarcón2 reported that 25% to 50% of patients with SLE had significant morbidity primarily from common bacterial infections, with viral, fungal, and parasitic infection less common. Staphylococcus aureus was a common cause of soft tissue infection, septic arthritis, and bacteremia. Streptococcus pneumoniae typically caused respiratory infections, although meningitis and sepsis were reported with SLE. Gram-negative bacteria such as Escherichia coli, Klebsiella species, and Pseudomonas species usually caused urinary tract infections and nosocomial pneumonia. Other bacterial infections included Nocardia species, Mycobacterium tuberculosis, and, rarely, Listeria monocytogenes. The most common viral infection was herpes zoster. Fungal infections included Pneumocystis jirovecii (formerly known as Pneumocystis carinii) and Candida species.
In scleroderma, another connective tissue disease evaluated in the literature by Alarcón,2 reports of bacterial, viral, and fungal infections are limited to case reports. In scleroderma patients, viral infections with cytomegalovirus (CMV), parvovirus B19, and P jirovecii were similar to pathogens observed with SLE.
In polymyositis/dermatomyositis, gram-positive pneumonia affected 15% to 20% of patients and S aureus occurred frequently in the juvenile form of the disease. Herpes zoster was commonly observed, but CMV was relatively rare. Other viral infections included Coxsackie virus, parvovirus B19, and hepatitis C in polymyositis/dermatomyositis. Infection with P jirovecii is frequently fatal in these patients. Other fungal infections seen in polymyositis/dermatomyositis include candidiasis and histoplasmosis.2
Since the approval of antitumor necrosis factor (anti-TNF) agents for RA in the late 1990s, as well as other more recent biologic agents, there has been heightened awareness of infectious complications in rheumatologic patients. A major concern with the anti-TNF agents is the risk of granulomatous infection, particularly mycobacterial disease and dimorphic fungal infections such as histoplasmosis and coccidioidomycosis. Formation of granulomas is the major host defense against mycobacterial infection and is mediated in large part by TNF-alpha. The precise risk of infection associated with each of the various biologic agents is still under study, and rates from randomized trials have differed from postmarketing surveillance studies. Important pathogens associated with biologic agents include Nocardia, CMV, Listeria, Aspergillus, and JC virus (JCV).3,4 Delays in the diagnosis of these infections in immunocompromised patients have led to poor outcomes.
KEY PATHOGENS IN INFECTIONS OF IMMUNOCOMPROMISED HOSTS
Pneumocystis jirovecii
For many decades, P jirovecii was classified as a protozoan but, based on gene sequencing, the organism has been reclassified as a fungus. P jirovecii is a low-virulence, unicellular organism that is the causative agent of Pneumocystis pneumonia (PCP). Epidemiologically, primary infection most likely occurs in infants and children. Colonization may be transient, entering the airways and then resolving over a period of weeks or months. Alternatively, the organism may enter a latent state similar to tuberculosis with reactivation occurring during times of intense immunosuppression. However, molecular epidemiology studies show that new cases of PCP are likely environmentally acquired through multiple exposures rather than reactivation of latent infection.5,6 Transmission is thought to be airborne from person to person. Pathogenically, the trophic form of the organism attaches to type 1 alveolar cells and remains in the extracellular compartment of the alveoli. This colonization evokes an influx of inflammatory cells (CD8 cells, neutrophils, and macrophages). However, not all colonizations result in pneumonia—even in advanced human immunodeficiency virus (HIV) infection. While there is an innate immunity through alveolar macrophages and pulmonary surfactant, alveolar macrophage response is impaired in HIV when the CD4 count is low. Cell-mediated immunity is the main defense against progression to pneumonia with assistance from costimulatory molecules (such as CD28 and CD2) as well as B cells.
Laboratory diagnosis. P jirovecii cannot be grown in culture for clinical purposes, and it is extremely difficult to culture even in the research setting. Cytologic stains such as the Wright-Giemsa and methamine silver stains are the mainstay of laboratory diagnosis. The yield for P jirovecii from routine expectorated sputum is very low and some laboratories discourage this approach. The sensitivity of nebulized sputum using hypertonic saline ranges from 50% to 90%.9
In patients with acquired immune deficiency syndrome (AIDS), bronchoscopy provides 90% to 98% sensitivity by BAL. Transbronchial biopsy may provide some additional yield over BAL in a few situations, such as patients who have been receiving partial P jirovecii prophylaxis. Immunofluorescence techniques using monoclonal antibodies to P jirovecii are commercially available and are first-line diagnostic tools in some laboratories. Recently, polymerase chain reaction (PCR) assay has been introduced into clinical practice as a reproducible test with high sensitivity.
Primary therapy. Primary therapy for PCP consists of trimethoprim-sulfamethoxazole (TMP-SMX) or pentamidine. TMP-SMX is considered the drug of choice and is usually administered intravenously for 21 days in HIV patients and 14 days for non-HIV patients. The oral form may be used in patients with less severe PCP with a functioning gastrointestinal tract. Common adverse reactions to TMP-SMX include rash, Stevens-Johnson syndrome, neutropenia, changes in pulmonary function, and nausea/vomiting/diarrhea.10 Pentamidine is as effective as TMP-SMX, but is associated with renal toxicity, hypotension, severe hypoglycemia, cardiac arrhythmias, and diabetes.11 It is generally reserved for severe cases of PCP in patients who are allergic to or otherwise intolerant of sulfa. Other treatments include atovaquone and trimethoprim-dapsone. Adjunctive corticosteroids have been shown to be beneficial in moderate to severe PCP in HIV patients to reduce the local host inflammatory response to dead or dying organisms. Recent guidelines have recommended corticosteroids for HIV patients with PCP who have an arterial oxygen pressure of 70 mm Hg or less on room air, or an alveolar-arterial (A-a) gradient of oxygen 35 mm Hg or greater.12 Little is known about the role of adjunctive corticosteroids in non-HIV patients, given a lack of clinical studies.
Prevention. Recent estimates of disease burden from a meta-analysis of 11,900 patients with connective tissue diseases found PCP in 12% of patients with GPA, in 6% of those with polydermatomyositis, in 5% of those with SLE, and in 1% of those with RA.1 Mortality due to PCP is higher in patients with rheumatic diseases, ranging from 30% in RA to 63% in GPA, than in those with HIV (10% to 20%).13 One key risk factor predisposing patients with connective tissue diseases to infection with P jirovecii is recent corticosteroid use. Among patients with connective tissue disease, more than 90% of those infected with P jirovecii have recently received steroid therapy.14 Additionally, in almost all patients with P jirovecii, lymphopenia with absolute lymphocyte counts less than 1,000/mm3 is present.15
In patients with HIV, prophylaxis is initiated at a CD4 level of 200/mm3.13 However, the cutoff is less clear for non-HIV rheumatic patients. A cutoff of less than 300 cells/mm3 has been proposed for prophylaxis of PCP. However, at that range, approximately 50% of patients with connective tissue disease would remain above the threshold.13 One possible solution is to screen by PCR and treat colonization. Other algorithms have been proposed, but there is no general consensus on treatment of non-HIV rheumatic patients.13,16 Generally, prophylaxis should be considered in patients at the highest risk for PCP. These include patients taking prednisone at doses greater than 20 mg/day for 1 month plus a cytotoxic agent, a TNF inhibitor plus glucocorticoids, and methotrexate plus glucocorticoids in GPA.13
Nocardia asteroides and Nocardia species
Classically, Nocardia infection results in abscess formation with infiltrates of polymorphonuclear cells, debris, and thin-walled abscesses. The most frequent site of primary infection is pulmonary. Characteristically, multiple pulmonary nodules or cavities are seen, and Nocardia should be considered in the differential diagnosis of an immunocompromised patient with nodular pneumonia. The nodules can also be masslike in appearance (greater than 2 cm). The presentation of new cavitary lung opacities with systemic symptoms may be mistaken for GPA.22Nocardia may disseminate to the central nervous system (CNS), skin, joints, and spine, usually causing suppurative infection at these sites. Nocardia has a very strong tropism for neural tissue. In the CNS, Nocardia can cause single or multiple brain abscesses that may be asymptomatic; patients with pulmonary nocardiosis require imaging to rule out occult CNS involvement.
Nocardia species are resistant to several antibiotics. The treatment of choice for Nocardia species is TMPSMX, but imipenem, amikacin, third-generation cephalosporins, and other options such as minocycline and linezolid may be considered depending on the species and the antimicrobial susceptibility pattern.
Histoplasma capsulatum
Histoplasma capsulatum is a dimorphic fungus that causes disease in both healthy and immunocompromised hosts. The organism differs from other pathogenic fungi in that it is an intracellular organism, mainly involving the reticuloendothelial system, and is rarely in the extracellular space. In the United States, infections are clustered endemically in areas such as the Mississippi and Ohio River Valleys, but infections are common worldwide. The fungus is found in soil, mulch, bird excrement, and bat guano. Asymptomatic or mild infections are common in healthy persons residing in endemic areas and occur on a sporadic basis. Epidemics can occur when contaminated material is aerosolized. Histoplasmosis is also an opportunistic infection in patients with impaired T-cell immunity such as persons with AIDS, organ transplant recipients, hematologic malignancies, and corticosteroid use. Clinically significant cases of histoplasmosis have been described in patients with RA while receiving methotrexate alone, corticosteroids alone, and combinations of disease-modifying agents.23 Histoplasmosis was recently identified in 240 patients in association with TNF inhibitors, translating to 17 per 100,000 patients treated with infliximab.21,24
Pathogenesis. Infection initially occurs through inhalation of contaminated material from the environment, primarily causing pulmonary infection. The organism converts from a mold form in the environment to a pathogenic yeast form in the host. Once inhaled, the mediastinal lymph nodes provide the first line of defense. Following draining of the lymph nodes, the organism enters the bloodstream in both immunocompetent and immunosuppressed patients. It is spread hematogenously into the spleen, liver, and reticulo-endothelial system, where it is eventually cleared. In immunocompetent patients, cellular immunity limits infection within 7 to 14 days and humoral immunity is not protective.25 Granuloma formation is the hallmark of host defense.
Spectrum of illness. Histoplasmosis is associated with a wide spectrum of illness, with presentation ranging from asymptomatic to mild pulmonary illness to overwhelming pneumonia. Symptomatic pulmonary histoplasmosis typically presents with fever, flulike symptoms, and cough, often with retrosternal chest pain. X-rays show patchy or nodular infiltrates, with hilar or mediastinal lymphadenopathy. In some cases the lung parenchyma is clear and the main feature is fever and bilateral hilar adenopathy. Pulmonary histoplasmosis may be difficult to distinguish from sarcoidosis and tuberculosis. Extrapulmonary disease can present as hepatitis, infective endocarditis, and chronic meningitis. In immunocompromised patients, histoplasmosis can present as a progressive disseminated disease which can be acute, subacute, or chronic. Chronic disseminated histoplasmosis is characterized by cough, persistent fever, wasting, hepatosplenomegaly, oral ulcerations, and progressive cytopenias. Acute disseminated histoplasmosis has a much more fulminant course characterized by respiratory insufficiency, hypotension, multisystem organ failure, coagulopathies, and encephalopathy. Histoplasmosis is primarily a pulmonary disease, but in disseminated disease more than 50% of patients have no pulmonary symptoms and 30% may have normal chest x-rays.26 In one series of infliximab-related cases (n = 10), all came from an endemic area 1 week to 6 months after the first dose of infliximab. Patients presented with cough, fever, and shortness of breath.27 The pathogenesis of histoplasmosis in patients receiving TNF inhibitors is not entirely clear; such patients may be suffering a new primary infection, a reinfection, or, least likely, reactivation of latent infection.
Definitive diagnosis requires culture confirmation from appropriate body fluids or identification of characteristic yeast forms from histopathologic sections of tissue biopsies. Serologic tests may also be used to confirm the diagnosis. Detection of H and M precipitins or bands by immunodiffusion is a routine test in many laboratories. M bands are present in 50% of acute cases but their presence does not distinguish acute from remote infection. H bands are present in only 10% of all acute cases, but their presence is very specific for acute histoplasmosis.28
When looking at complement fixation antibodies to yeast (HY) and mycelial (HMy) forms in pulmonary histoplasmosis, a fourfold rise in titer establishes the diagnosis retrospectively, and a single titer greater than 1:32 is strongly suggestive of active infection. However, in progressive disseminated histoplasmosis, the complement fixation antibodies are frequently negative.29 Detection of antigen in urine and serum by enzyme immunoassay has become a mainstay of diagnosis, with a sensitivity of approximately 90% in progressive disseminated disease.30 Of note, most cases of histoplasmosis associated with biologic agents have detectable urinary antigen tests.
Treatment. Acute pulmonary histoplasmosis is usually self-limited, requiring no treatment. The 2007 Infective Diseases Society of America (IDSA) guidelines recommend observation alone in most cases of mild to moderate pulmonary histoplasmosis unless symptoms persist longer than 1 month. For moderately severe or severe acute pulmonary histoplasmosis, the IDSA recommends lipid formulations of amphotericin B (3.0 to 5.0 mg/kg/day) or deosycholate amphotericin B (0.7 to 1.0 mg/kg/day) for 1 to 2 weeks followed by itraconazole 200 mg twice daily for a total of 12 weeks. Methylprednisolone at a dose of 0.5 to 1.0 mg/kg/day intravenously for 1 to 2 weeks is also recommended. For moderately severe to severe disseminated histoplasmosis, the IDSA recommends lipid formulations of amphotericin B (3.0 mg/kg/day) for 1 to 2 weeks followed by oral itraconazole 200 mg three times daily for 3 days and then 200 mg twice daily for a total of at least 12 months.31 Commonly, the immunosuppressive agent is held during treatment.
Aspergillus species
Another emerging pathogen is Aspergillus species—a ubiquitous mold spread by aerosols of spores. There are many different species of Aspergillus, but the most common human pathogens include A fumigates, A niger, and A flavus. To date, 39 cases of Aspergillus infection associated with infliximab and etanercept have been reported in the Adverse Event Reporting System, translating to 9 to 12 cases per 100,000 patients.21
Varicella zoster
JC virus
More than 80% of adults are seropositive for JCV, a DNA virus of the genus Polyomavirus that causes lytic infection of oligodendrocytes.34 In immunocompromised hosts, JCV causes progressive multifocal leukoencephalopathy (PML), a rare but devastating demyelinating disease. PML was first described in malignancy, leukemia, and various other immunocompromised states, prior to its strong association with AIDS in the 1980s. More recently, JCV has been associated with natalizumab for multiple sclerosis and Crohn disease, rituximab for oncology patients, efalizumab for psoriasis,35 and mycophenolate mofetil for transplant recipients.36
In 2006 the US Food and Drug Administration issued a safety alert regarding PML in two patients with SLE treated with rituximab and other immunosuppressives.37 In a review of PML in rheumatic disease, 36 cases were identified in patients who had not previously received a biologic agent. Most of these patients (60%) had SLE.38 Of these, many had little or no immunosuppression over the 6 months prior to the diagnosis of PML, suggesting that SLE itself may predispose to PML. Interestingly, PML is rarely associated with TNF inhibitors.
Classic presentation of PML includes motor weakness, aphasia, dysarthria, vision loss, and cognitive loss. Atypical presentation includes seizures, headaches, and brainstem involvement. PML usually spares the optic nerves, spinal cord, peripheral nerves, and muscles. In persons with underlying rheumatic diseases, PML can be difficult to distinguish from neuropsychiatric SLE or CNS vasculitis.
Treatment. In clinical trials no antiviral agent has been effective in the treatment of PML. In HIV patients who develop PML, highly active antiretroviral therapy should be initiated (if antiretroviral-naïve) or existing antiviral regimens optimized. Antiretroviral therapy in this situation may stabilize disease and possibly increase survival.42 For HIV-negative patients who develop PML, the cornerstone of management is immediate decrease or discontinuation of immunosuppression.43 Several adjunctive measures have been reported mainly in natalizumab-associated PML, including corticosteroids, mirtazapine, plasma exchange, and others.
VACCINES
Vaccination is important in the prevention of infectious disease in immunocompromised patients with connective tissue diseases. Because live vaccines are contraindicated in immunocompromised patients, inactivated or component vaccines should be used. It is recommended that patients who will start immunosuppressive therapy be vaccinated 2 to 4 weeks before beginning therapy. If this is not possible, vaccination should be administered during disease remission, 3 months after immunosuppression and 1 to 3 months after administration of high-dose corticosteroids.
- Short-term (less than 14 days)
- At a dose of less than 20 mg/day of prednisone or equivalent
- Long-term on alternate days with short-acting preparations
- At a physiologic dose of prednisone
- Topical, inhaled, intra-articular, bursal, or via tendon.44
Until definitive guidelines are developed, practitioners must evaluate and treat each patient individually to maximize the efficacy of disease treatments while preventing infection morbidity and mortality in their patients with connective tissue diseases.
- Falagas ME, Manta KG, Betsi GI, Pappas G. Infection-related morbidity and mortality in patients with connective tissue diseases: a systematic review. Clin Rheumatol 2007; 26:663–670.
- Alarcón GS. Infections in systemic connective tissue diseases: systemic lupus erythematosus, scleroderma, and polymyositis/dermatomyositis. Infect Dis Clin North Am 2006; 20:849–875.
- Crum NF, Lederman ER, Wallace MR. Infections associated with tumor necrosis factor-alpha antagonists. Medicine (Baltimore) 2005; 84:291–302.
- Rychly DJ, DiPiro JT. Infections associated with tumor necrosis factor-alpha antagonists. Pharmacotherapy 2005; 25:1181–1192.
- Wakefield AE, Lindley AR, Ambrose HE, Denis CM, Miller RF. Limited asymptomatic carriage of Pneumocystis jiroveci in human immunodeficiency virus-infected patients [published online ahead of print March 6, 2003]. J Infect Dis 2003; 187:901–908. doi: 10.1086/368165
- Beard CB, Carter JL, Keely SP, et al. Genetic variation in Pneumocystis carinii isolates from different geographic regions: implications for transmission. Emerg Infect Dis 2000; 6:265–272.
- Walzer PD, Smulian AG. Pneumocystis species. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, Pa: Elsevier Churchill Livingstone; 2009.
- Hartman TE, Primack SL, Müller NL, Staples CA. Diagnosis of thoracic complications in AIDS: accuracy of CT. Am J Roentgenol 1994; 162:547–553.
- Shelhamer JH, Gill VJ, Quinn TC, et al. The laboratory evaluation of opportunistic pulmonary infections. Ann Intern Med 1996; 124:585–599.
- Wharton JM, Coleman DL, Wofsy CB, et al. Trimethoprim-sulfamethoxazole or pentamidine for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. Ann Intern Med 1986; 105:37–44.
- Stein DS, Stevens RC. Treatment-associated toxicities: incidence and mechanisms. In:Sattler FR, Walzer PD, eds. Pneumocystis carinii. London: Bailliere Tindall; 1995:505–530.
- Consensus statement on the use of corticosteroids as adjunctive therapy for Pneumocystis pneumonia in the acquired immunodeficiency syndrome. The National Institutes of Health-University of California Expert Panel for Corticosteroids as Adjunctive Therapy for Pneumocystis Pneumonia. N Engl J Med 1990; 323:1500–1504.
- Stamp LK, Hurst M. Is there a role for consensus guidelines for P. jiroveci pneumonia prophylaxis in immunosuppressed patients with rheumatic diseases? J Rheumatol 2010; 37:686–688.
- Yale S, Limper A. Pneumocystis carinii pneumonia in patients without acquired immunodeficiency syndrome: associated illnesses and prior corticosteroid therapy. Mayo Clin Proc 1996; 71:5–13.
- Sowden E, Carmichael A. Autoimmune inflammatory disorders, systemic corticosteroids and Pneumocystis pneumonia: a strategy for prevention [published online October 16, 2004]. BMC Infect Dis 2004; 4:42. doi: 10.1186/1471-2334-4-42
- Cettomai D, Gelber AC, Christopher-Stine L. A survey of rheumatologists’ practice for prescribing Pneumocystis prophylaxis. J Rheumatol 2010; 37:792–799.
- Keegan JM, Byrd JW. Nocardiosis associated with low dose methotrexate for rheumatoid arthritis. J Rheumatol 1988; 15:1585–1586.
- Gruberg L, Thaler M, Rozenman J, et al. Nocardia asteroides infection complicating rheumatoid arthritis. J Rheumatol 1991; 18:459–461.
- Corneliessen JJ, Bakker LJ, van der Veen MJ, et al. Nocardia asteroides pneumonia complicating low dose methotrexate treatment of refractory rheumatoid arthritis. Ann Rheum Dis 1991; 50;642–644.
- Silva C, Faccioli LH. Tumor necrosis factor and macrophage activation are important in clearance of Nocardia brasiliensis from the livers and spleens of mice. Infect Immun 1992; 60:3566–3570.
- Wallis RS, Broder MS, Wong JY, Hanson ME, Beenhouwer DO. Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin Infect Dis 2004; 38:1261–1265.
- Gibb W, Williams A. Nocardiosis mimicking Wegener’s granulomatosis. Scand J Infect Dis 1986; 18:583–585.
- Olson TC, Bongartz T, Crowson CS, Roberts GD, Orenstein R, Matteson EI. Histoplasmosis infection in patients with rheumatoid arthritis, 1998–2009 [published online May 23, 2011]. BMC Infectious Diseases 2011; 11:145. doi: 10.1186/1471-2334-11-145
- Information for Healthcare Professionals: Cimzia (certolizumab pegol), Enbrel etanercept), Humira (adalimumab), and Remicade (infliximab). U.S. Food and Drug Administration Web site. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124185.htm. Updated January 25, 2010. Accessed September 27, 2012.
- Paya CV, Roberts GD, Cockerill FR. Transient fungemia in acute pulmonary histoplasmosis: detection by new blood-culturing techniques. J Infect Dis 1987; 156:313–315.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Lee JH, Slifman NR, Gershon SK, et al. Life-threatening histoplasmosis complicating immunotherapy with tumor necrosis factor alpha antagonists infliximab and etanercept. Arthritis Rheum 2002; 46:2565–2570.
- Picardi JL, Kauffman CA, Schwarz J, Phair JP. Detection of precipitating antibodies to Histoplasma capsulatum by counterimmunoelectrophoresis. Am Rev Respir Dis 1976; 114:171–176.
- Deepe GS. Histoplasma capsulatum. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, Pa: Elsevier Churchill Livingstone; 2009.
- Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev 2007; 20:115–132.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America [published online ahead of print August 27, 2007]. Clin Infect Dis 2007; 45:807–825. doi: 10.1086/521259
- Johnson RW. Herpes zoster and postherpetic neuralgia. Expert Rev Vaccines 2010; 9( 3 suppl):21–26.
- Wolfe F, Michaud K, Chakravarty EF. Rates and predictors of herpes zoster in patients with rheumatoid arthritis and non-inflammatory musculoskeletal disorders. Rheumatology 2006; 45:1370–1375.
- Weber T, Trebst C, Frye S, et al. Analysis of the systemic and intrathecal humoral immune response in progressive multifocal leukoencephalopathy. J Infect Dis 1997; 176:250–254.
- Kothary N, Diak IL, Brinker A, Bezabeh S, Avigan M, Dal Pan G. Progressive multifocal leukoencephalopathy associated with efalizumab use in psoriasis patients. J Am Acad Dermatol 2011; 65:546–551.
- Neff RT, Hurst FP, Falta EM, et al. Progressive multifocal leukoencephalopathy and use of mycophenolate mofetil after kidney transplantation. Transplantation 2008; 86:1474–1478.
- Rituxan warning. FDA Consum 2007; 41:3.
- Calabrese LH, Molloy ES, Huang D, Ransohoff RM. Progressive multifocal leukoencephalopathy in rheumatic diseases. Arthritis Rheum 2007; 56:2116–2128.
- Cinque P, Scarpellini P, Vago L, Linde A, Lazzarin A. Diagnosis of central nervous system complications in HIV-infected patients: cerebrospinal fluid analysis by the polymerase chain reaction. AIDS 1997; 11:1–17.
- Ryschkewitsch C, Jensen P, Hou J, Fahle G, Fischer S, Major EO. Comparison of PCR-southern hybridization and quantitative real-time PCR for the detection of JC and BK viral nucleotide sequences in urine and cerebrospinal fluid. J Virol Methods 2004; 121:217–221.
- Major EO. History and current concepts in the pathogenesis of PML. Cleve Clin J Med 2011; 78( suppl 2):S3–S7.
- Antinori A, Ammassari A, Giancola ML, et al. Epidemiology and prognosis of AIDS-associated progressive multifocal leukoencephalopathy in the HAART era. J Neurovirol 2001; 7:323–328.
- Calabrese L. A rational approach to PML for the clinician. Cleve Clin J Med 2011; 78 (suppl 2):S38–S41.
- Kroger AT, Sumaya CV, Pickering LK, Atkinson WL. General recommendations on immunization. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2011; 60:1–60.
- Falagas ME, Manta KG, Betsi GI, Pappas G. Infection-related morbidity and mortality in patients with connective tissue diseases: a systematic review. Clin Rheumatol 2007; 26:663–670.
- Alarcón GS. Infections in systemic connective tissue diseases: systemic lupus erythematosus, scleroderma, and polymyositis/dermatomyositis. Infect Dis Clin North Am 2006; 20:849–875.
- Crum NF, Lederman ER, Wallace MR. Infections associated with tumor necrosis factor-alpha antagonists. Medicine (Baltimore) 2005; 84:291–302.
- Rychly DJ, DiPiro JT. Infections associated with tumor necrosis factor-alpha antagonists. Pharmacotherapy 2005; 25:1181–1192.
- Wakefield AE, Lindley AR, Ambrose HE, Denis CM, Miller RF. Limited asymptomatic carriage of Pneumocystis jiroveci in human immunodeficiency virus-infected patients [published online ahead of print March 6, 2003]. J Infect Dis 2003; 187:901–908. doi: 10.1086/368165
- Beard CB, Carter JL, Keely SP, et al. Genetic variation in Pneumocystis carinii isolates from different geographic regions: implications for transmission. Emerg Infect Dis 2000; 6:265–272.
- Walzer PD, Smulian AG. Pneumocystis species. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, Pa: Elsevier Churchill Livingstone; 2009.
- Hartman TE, Primack SL, Müller NL, Staples CA. Diagnosis of thoracic complications in AIDS: accuracy of CT. Am J Roentgenol 1994; 162:547–553.
- Shelhamer JH, Gill VJ, Quinn TC, et al. The laboratory evaluation of opportunistic pulmonary infections. Ann Intern Med 1996; 124:585–599.
- Wharton JM, Coleman DL, Wofsy CB, et al. Trimethoprim-sulfamethoxazole or pentamidine for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. Ann Intern Med 1986; 105:37–44.
- Stein DS, Stevens RC. Treatment-associated toxicities: incidence and mechanisms. In:Sattler FR, Walzer PD, eds. Pneumocystis carinii. London: Bailliere Tindall; 1995:505–530.
- Consensus statement on the use of corticosteroids as adjunctive therapy for Pneumocystis pneumonia in the acquired immunodeficiency syndrome. The National Institutes of Health-University of California Expert Panel for Corticosteroids as Adjunctive Therapy for Pneumocystis Pneumonia. N Engl J Med 1990; 323:1500–1504.
- Stamp LK, Hurst M. Is there a role for consensus guidelines for P. jiroveci pneumonia prophylaxis in immunosuppressed patients with rheumatic diseases? J Rheumatol 2010; 37:686–688.
- Yale S, Limper A. Pneumocystis carinii pneumonia in patients without acquired immunodeficiency syndrome: associated illnesses and prior corticosteroid therapy. Mayo Clin Proc 1996; 71:5–13.
- Sowden E, Carmichael A. Autoimmune inflammatory disorders, systemic corticosteroids and Pneumocystis pneumonia: a strategy for prevention [published online October 16, 2004]. BMC Infect Dis 2004; 4:42. doi: 10.1186/1471-2334-4-42
- Cettomai D, Gelber AC, Christopher-Stine L. A survey of rheumatologists’ practice for prescribing Pneumocystis prophylaxis. J Rheumatol 2010; 37:792–799.
- Keegan JM, Byrd JW. Nocardiosis associated with low dose methotrexate for rheumatoid arthritis. J Rheumatol 1988; 15:1585–1586.
- Gruberg L, Thaler M, Rozenman J, et al. Nocardia asteroides infection complicating rheumatoid arthritis. J Rheumatol 1991; 18:459–461.
- Corneliessen JJ, Bakker LJ, van der Veen MJ, et al. Nocardia asteroides pneumonia complicating low dose methotrexate treatment of refractory rheumatoid arthritis. Ann Rheum Dis 1991; 50;642–644.
- Silva C, Faccioli LH. Tumor necrosis factor and macrophage activation are important in clearance of Nocardia brasiliensis from the livers and spleens of mice. Infect Immun 1992; 60:3566–3570.
- Wallis RS, Broder MS, Wong JY, Hanson ME, Beenhouwer DO. Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin Infect Dis 2004; 38:1261–1265.
- Gibb W, Williams A. Nocardiosis mimicking Wegener’s granulomatosis. Scand J Infect Dis 1986; 18:583–585.
- Olson TC, Bongartz T, Crowson CS, Roberts GD, Orenstein R, Matteson EI. Histoplasmosis infection in patients with rheumatoid arthritis, 1998–2009 [published online May 23, 2011]. BMC Infectious Diseases 2011; 11:145. doi: 10.1186/1471-2334-11-145
- Information for Healthcare Professionals: Cimzia (certolizumab pegol), Enbrel etanercept), Humira (adalimumab), and Remicade (infliximab). U.S. Food and Drug Administration Web site. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124185.htm. Updated January 25, 2010. Accessed September 27, 2012.
- Paya CV, Roberts GD, Cockerill FR. Transient fungemia in acute pulmonary histoplasmosis: detection by new blood-culturing techniques. J Infect Dis 1987; 156:313–315.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Lee JH, Slifman NR, Gershon SK, et al. Life-threatening histoplasmosis complicating immunotherapy with tumor necrosis factor alpha antagonists infliximab and etanercept. Arthritis Rheum 2002; 46:2565–2570.
- Picardi JL, Kauffman CA, Schwarz J, Phair JP. Detection of precipitating antibodies to Histoplasma capsulatum by counterimmunoelectrophoresis. Am Rev Respir Dis 1976; 114:171–176.
- Deepe GS. Histoplasma capsulatum. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, Pa: Elsevier Churchill Livingstone; 2009.
- Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev 2007; 20:115–132.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America [published online ahead of print August 27, 2007]. Clin Infect Dis 2007; 45:807–825. doi: 10.1086/521259
- Johnson RW. Herpes zoster and postherpetic neuralgia. Expert Rev Vaccines 2010; 9( 3 suppl):21–26.
- Wolfe F, Michaud K, Chakravarty EF. Rates and predictors of herpes zoster in patients with rheumatoid arthritis and non-inflammatory musculoskeletal disorders. Rheumatology 2006; 45:1370–1375.
- Weber T, Trebst C, Frye S, et al. Analysis of the systemic and intrathecal humoral immune response in progressive multifocal leukoencephalopathy. J Infect Dis 1997; 176:250–254.
- Kothary N, Diak IL, Brinker A, Bezabeh S, Avigan M, Dal Pan G. Progressive multifocal leukoencephalopathy associated with efalizumab use in psoriasis patients. J Am Acad Dermatol 2011; 65:546–551.
- Neff RT, Hurst FP, Falta EM, et al. Progressive multifocal leukoencephalopathy and use of mycophenolate mofetil after kidney transplantation. Transplantation 2008; 86:1474–1478.
- Rituxan warning. FDA Consum 2007; 41:3.
- Calabrese LH, Molloy ES, Huang D, Ransohoff RM. Progressive multifocal leukoencephalopathy in rheumatic diseases. Arthritis Rheum 2007; 56:2116–2128.
- Cinque P, Scarpellini P, Vago L, Linde A, Lazzarin A. Diagnosis of central nervous system complications in HIV-infected patients: cerebrospinal fluid analysis by the polymerase chain reaction. AIDS 1997; 11:1–17.
- Ryschkewitsch C, Jensen P, Hou J, Fahle G, Fischer S, Major EO. Comparison of PCR-southern hybridization and quantitative real-time PCR for the detection of JC and BK viral nucleotide sequences in urine and cerebrospinal fluid. J Virol Methods 2004; 121:217–221.
- Major EO. History and current concepts in the pathogenesis of PML. Cleve Clin J Med 2011; 78( suppl 2):S3–S7.
- Antinori A, Ammassari A, Giancola ML, et al. Epidemiology and prognosis of AIDS-associated progressive multifocal leukoencephalopathy in the HAART era. J Neurovirol 2001; 7:323–328.
- Calabrese L. A rational approach to PML for the clinician. Cleve Clin J Med 2011; 78 (suppl 2):S38–S41.
- Kroger AT, Sumaya CV, Pickering LK, Atkinson WL. General recommendations on immunization. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2011; 60:1–60.