User login
Our remarkable progress in understanding progressive multifocal leukoencephalopathy (PML) since its discovery more than 50 years ago has evolved in three stages, concurrent with the changing epidemiology of PML: the pre–human immunodeficiency virus (HIV) era; the HIV era, with highly active antiretroviral therapy (HAART) bringing further change; and the biologic therapy era.
Before the appearance of HIV, PML developed mostly in patients who had lymphoma, other malignancies, and rare forms of immunosuppression. The development of HIV changed the nature of PML, with more than 75% of cases now reported in HIV-infected patients. Within the HIV population, the epidemiology and prognosis of PML have undergone additional changes since the late 1990s. The introduction of HAART transformed PML from an almost uniformly fatal and inexorably progressive disease to one in which long-term survival is expected, particularly in the setting of robust immune reconstitution.1
The third and most recent stage in the evolution of PML and our understanding of it has coincided with the introduction and use of increasingly potent immunosuppressive regimens and novel biologic immunologic therapies that target various aspects of the integrated immune response. These agents are being applied not only in the field of autoimmune and autoinflammatory disease but also in transplantation and oncology.
Collectively, vulnerable populations (ie, patients with lymphoreticular malignancies and autoinflammatory diseases) are now being subjected to therapies that singly or in combination have unknown effects on the immune system. As a byproduct, practitioners who were only vaguely aware of PML in the past now must consider PML in their differential diagnosis, develop a rational plan for evaluating such patients, and recognize when referral to a specialist is indicated. Recent descriptions of atypical forms of PML2,3 add to the challenge for clinicians, as do reports of cases of PML in patients with minimal immunosuppression, in the absence of immunosuppressive therapy, and in patients who appear to have “normal” immune systems but in fact have diseases such as sarcoidosis.4 Rare cases are also being reported in patients with advanced liver disease.4
This article offers recommendations for enhanced awareness of PML, suggestions for improved evaluation of predisposed patients, and a summary of currently accepted treatment strategies.
WHEN TO SUSPECT PML

Patients being treated with immunosuppressive biologic agents represent a significant group that is predisposed to PML. At one time, focal neurologic deficits were required to consider the possibility of PML, but cognitive/behavioral abnormalities rather than focal neurologic findings are often the presenting sign in individuals treated with immune-modulating biologic agents. This phenomenon is most strikingly observed in recipients of natalizumab. Any central nervous system (CNS) dysfunction in a patient taking an immunosuppressive biologic agent should arouse suspicion for PML.
Peripheral neuropathy is not caused by PML but can coexist with it. Accordingly, in patients with rheumatologic disease who are receiving immune modifiers, neuromuscular symptoms in the absence of brain abnormalities on magnetic resonance imaging (MRI) argue against consideration of PML but do not rule it out—especially in patients with connective tissue diseases.
Systemic lupus erythematosus (SLE) represents a special challenge for several reasons. First, SLE appears to be a predisposing factor among other connective tissue diseases.5 In addition, SLE is associated with a variety of CNS complications, including a spectrum of focal and diffuse signs and symptoms that can mimic PML and lead to underdiagnosis.
Underrecognition is a risk in the HIV population as well, where cognitive impairment is common. Irrespective of immune or virologic status, 57% of HIV patients demonstrate impairment on neuropsychiatric testing. Often, mild to moderate cognitive impairment in HIV is attributed to HIV encephalopathy with no further workup, resulting in a missed or late diagnosis of PML.
IMAGING CONSIDERATIONS
In the rheumatologic disease population, especially those with SLE, and the HIV population, neuroimaging is indicated in any patient who presents with cognitive impairment. Typical radiographic characteristics of PML on MRI are subcortical white matter hyperintense areas on T2-weighted images and fluid-attenuated inversion recovery. T1-weighted images will reveal hypointense lesions that usually do not enhance, but may do so in fewer than 10% of patients with PML. Typically, no mass effect is seen.
In addition to rare faint gadolinium enhancement of lesions, other lesion characteristics may depart from the classic picture—for example, white matter and gray matter involvement, and monofocal instead of multifocal lesions. In HIV-positive patients, MRI can demonstrate diffuse cerebellar atrophy and subtle white matter abnormalities within the cerebellum.
Unfortunately, nonspecific white matter lesions occur in HIV infection as well as connective tissue diseases, compromising diagnostic specificity of a single imaging study. Nevertheless, progression of clinical signs and symptoms and progressive MRI changes should prompt a more vigorous diagnostic evaluation for PML. Alternatively, a normal MRI in a patient in whom PML is suspected has strong negative predictive value. In either situation, baseline neuroimaging is not recommended.
DIAGNOSIS AND REFERRAL
A neurology consult is advised when a patient has a predisposing condition for PML or suspicious neurologic signs or symptoms, whether focal or diffuse, and in whom an MRI demonstrates white matter changes.
Evaluation for JC virus DNA
When the neurology consult has been scheduled, and before the actual visit to the neurologist, a cerebrospinal fluid (CSF) sample should be obtained and evaluated for JC virus (JCV) DNA using a highly sensitive polymerase chain reaction (PCR) assay. Lumbar puncture in the setting of possible PML is critical to exclude the presence of other opportunistic infections.
The importance of using ultrasensitive PCR assays for diagnosing PML cannot be overstated, as falsely negative CSF PCR has been observed for JCV DNA despite high levels of JCV DNA in spinal fluid when utilizing less sensitive assays. The most sensitive commercial assays can detect as few as 50 copies of JCV DNA per mL of CSF fluid.
The risk of PML imparted by biologic agents other than natalizumab and nonbiologic immunosuppressive agents has been difficult to quantify, but no immune-modifying drug or combination of drugs appears entirely free of risk. Any patient who has had significant or prolonged immunosuppression should be considered vulnerable, and any patient with suspicion of PML based on unexplained neurologic symptoms warrants CSF examination for JCV DNA.
Brain biopsy
In patients with progressive clinical and MRI findings that suggest PML, but whose CSF PCR for JCV DNA is repeatedly negative, a brain biopsy is appropriate regardless of background immunosuppression. In the patient with rheumatologic disease, for example, suspicion of PML should be heightened if there is neurologic deterioration in the face of escalating antiinflammatory or immunosuppressive therapy for immune-driven inflammatory disease. Diagnostic urgency is particularly warranted in those disorders where the possibility for immune reconstitution is highest (ie, those receiving immunosuppressive regimens).
PML MANAGEMENT DEPENDS ON CLINICAL SETTING
Management of PML starts with risk stratification to identify those patients most prone to developing PML based on their immune status; the presence of autoimmune disease; the subtype of disease (in the case of SLE); and the nature, intensity, and duration of their immunosuppression. If a high-risk patient develops signs and symptoms of PML, the diagnosis should be anticipated and serious consideration given to withholding immunosuppressive therapies while the patient is being worked up.
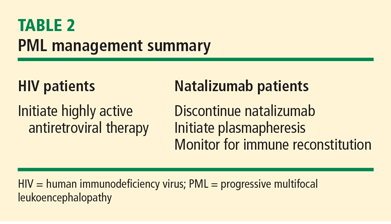
Accelerating immune reconstitution
Once a diagnosis of PML is confirmed, immune reconstitution should be accelerated whenever possible. This can include temporary or permanent withdrawal of immunosuppressive therapy and initiation of plasmapheresis. Evidence supports continuing plasma exchange until natalizumab serum drug levels decline to less than 1 μg/mL to achieve desaturation of the alpha-4 integrin receptor.6 Typically, desaturation of the targeted integrin receptor occurs after five plasmapheresis sessions.
Immune reconstitution may also precipitate a syndrome known as the immune reconstitution inflammatory syndrome (IRIS), characterized by enlargement and contrast enhancement of PML lesions, appearance of new brain lesions, and worsening of neurologic deficits. The infiltration of the brain with inflammatory multinucleated cells and lymphocytes following abrupt immune reconstitution requires treatment. Opinion suggests that judicious use of corticosteroids may control the immune response in the brain in patients with PML-IRIS,7,8 although further studies are needed.
Involving the patient in treatment decisions
Because risk tolerance varies considerably among individuals, patients should be informed of the risks of PML on the basis of their disease and the agents used to treat it. They should also be given information about the effects of individual treatments on the course of their disease, and they should be encouraged to participate in the selection of therapy.
SUMMARY
The approach to PML in the biologic era starts with an increased awareness of the disease followed by recognition of vulnerable populations and factors that contribute to the development of PML, such as biologic and nonbiologic immunosuppressive therapy. Optimal management includes a low threshold for investigating neurologic signs and symptoms and new-onset signs and symptoms in vulnerable populations, the use of MRI to detect typical PML brain lesions and other atypical brain features (ie, cerebellar atrophy), lumbar puncture and spinal CSF analysis to detect JCV DNA, and timely neurologic consultation for further evaluation. Much still needs to be learned about PML and the risks imparted by background diseases and individual drugs used in rheumatologic, neurologic, and oncologic disease.
- Brew BJ, Davies NW, Cinque P, Clifford DB, Nath A. Progressive multifocal leukoencephalopathy and other forms of JC virus disease. Nat Rev Neurol 2010; 6:667–679.
- Kedar S, Berger JR. The changing landscape of progressive multifocal leukoencephalopathy. Curr Infect Dis Rep 2011; 13:380–386.
- Tan CS, Koralnik IJ. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol 2010; 9:425–437.
- Gheuens S, Pierone G, Peeters P, Koralnik IJ. Progressive multifocal leukoencephalopathy in individuals with minimal or occult immunosuppression. J Neurol Neurosurg Psychiatry 2010; 81:247–254.
- Molloy ES, Calabrese LH. Progressive multifocal leukoencephalopathy: a national estimate of frequency in systemic lupus erythematosus and other rheumatic diseases. Arthritis Rheum 2009; 60:3761–3765.
- Khatri BO, Man S, Giovannoni G, et al. Effect of plasma exchange in accelerating natalizumab clearance and restoring leukocyte function. Neurology 2009; 72:402–409.
- Clifford DB, De Luca A, Simpson DM, Arendt G, Giovannoni G, Nath A. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol 2010; 9:438–446.
- Johnson T, Nath A. Immune reconstitution inflammatory syndrome and the central nervous system. Curr Opin Neurol 2011; 24:284–290.
Our remarkable progress in understanding progressive multifocal leukoencephalopathy (PML) since its discovery more than 50 years ago has evolved in three stages, concurrent with the changing epidemiology of PML: the pre–human immunodeficiency virus (HIV) era; the HIV era, with highly active antiretroviral therapy (HAART) bringing further change; and the biologic therapy era.
Before the appearance of HIV, PML developed mostly in patients who had lymphoma, other malignancies, and rare forms of immunosuppression. The development of HIV changed the nature of PML, with more than 75% of cases now reported in HIV-infected patients. Within the HIV population, the epidemiology and prognosis of PML have undergone additional changes since the late 1990s. The introduction of HAART transformed PML from an almost uniformly fatal and inexorably progressive disease to one in which long-term survival is expected, particularly in the setting of robust immune reconstitution.1
The third and most recent stage in the evolution of PML and our understanding of it has coincided with the introduction and use of increasingly potent immunosuppressive regimens and novel biologic immunologic therapies that target various aspects of the integrated immune response. These agents are being applied not only in the field of autoimmune and autoinflammatory disease but also in transplantation and oncology.
Collectively, vulnerable populations (ie, patients with lymphoreticular malignancies and autoinflammatory diseases) are now being subjected to therapies that singly or in combination have unknown effects on the immune system. As a byproduct, practitioners who were only vaguely aware of PML in the past now must consider PML in their differential diagnosis, develop a rational plan for evaluating such patients, and recognize when referral to a specialist is indicated. Recent descriptions of atypical forms of PML2,3 add to the challenge for clinicians, as do reports of cases of PML in patients with minimal immunosuppression, in the absence of immunosuppressive therapy, and in patients who appear to have “normal” immune systems but in fact have diseases such as sarcoidosis.4 Rare cases are also being reported in patients with advanced liver disease.4
This article offers recommendations for enhanced awareness of PML, suggestions for improved evaluation of predisposed patients, and a summary of currently accepted treatment strategies.
WHEN TO SUSPECT PML
Patients being treated with immunosuppressive biologic agents represent a significant group that is predisposed to PML. At one time, focal neurologic deficits were required to consider the possibility of PML, but cognitive/behavioral abnormalities rather than focal neurologic findings are often the presenting sign in individuals treated with immune-modulating biologic agents. This phenomenon is most strikingly observed in recipients of natalizumab. Any central nervous system (CNS) dysfunction in a patient taking an immunosuppressive biologic agent should arouse suspicion for PML.
Peripheral neuropathy is not caused by PML but can coexist with it. Accordingly, in patients with rheumatologic disease who are receiving immune modifiers, neuromuscular symptoms in the absence of brain abnormalities on magnetic resonance imaging (MRI) argue against consideration of PML but do not rule it out—especially in patients with connective tissue diseases.
Systemic lupus erythematosus (SLE) represents a special challenge for several reasons. First, SLE appears to be a predisposing factor among other connective tissue diseases.5 In addition, SLE is associated with a variety of CNS complications, including a spectrum of focal and diffuse signs and symptoms that can mimic PML and lead to underdiagnosis.
Underrecognition is a risk in the HIV population as well, where cognitive impairment is common. Irrespective of immune or virologic status, 57% of HIV patients demonstrate impairment on neuropsychiatric testing. Often, mild to moderate cognitive impairment in HIV is attributed to HIV encephalopathy with no further workup, resulting in a missed or late diagnosis of PML.
IMAGING CONSIDERATIONS
In the rheumatologic disease population, especially those with SLE, and the HIV population, neuroimaging is indicated in any patient who presents with cognitive impairment. Typical radiographic characteristics of PML on MRI are subcortical white matter hyperintense areas on T2-weighted images and fluid-attenuated inversion recovery. T1-weighted images will reveal hypointense lesions that usually do not enhance, but may do so in fewer than 10% of patients with PML. Typically, no mass effect is seen.
In addition to rare faint gadolinium enhancement of lesions, other lesion characteristics may depart from the classic picture—for example, white matter and gray matter involvement, and monofocal instead of multifocal lesions. In HIV-positive patients, MRI can demonstrate diffuse cerebellar atrophy and subtle white matter abnormalities within the cerebellum.
Unfortunately, nonspecific white matter lesions occur in HIV infection as well as connective tissue diseases, compromising diagnostic specificity of a single imaging study. Nevertheless, progression of clinical signs and symptoms and progressive MRI changes should prompt a more vigorous diagnostic evaluation for PML. Alternatively, a normal MRI in a patient in whom PML is suspected has strong negative predictive value. In either situation, baseline neuroimaging is not recommended.
DIAGNOSIS AND REFERRAL
A neurology consult is advised when a patient has a predisposing condition for PML or suspicious neurologic signs or symptoms, whether focal or diffuse, and in whom an MRI demonstrates white matter changes.
Evaluation for JC virus DNA
When the neurology consult has been scheduled, and before the actual visit to the neurologist, a cerebrospinal fluid (CSF) sample should be obtained and evaluated for JC virus (JCV) DNA using a highly sensitive polymerase chain reaction (PCR) assay. Lumbar puncture in the setting of possible PML is critical to exclude the presence of other opportunistic infections.
The importance of using ultrasensitive PCR assays for diagnosing PML cannot be overstated, as falsely negative CSF PCR has been observed for JCV DNA despite high levels of JCV DNA in spinal fluid when utilizing less sensitive assays. The most sensitive commercial assays can detect as few as 50 copies of JCV DNA per mL of CSF fluid.
The risk of PML imparted by biologic agents other than natalizumab and nonbiologic immunosuppressive agents has been difficult to quantify, but no immune-modifying drug or combination of drugs appears entirely free of risk. Any patient who has had significant or prolonged immunosuppression should be considered vulnerable, and any patient with suspicion of PML based on unexplained neurologic symptoms warrants CSF examination for JCV DNA.
Brain biopsy
In patients with progressive clinical and MRI findings that suggest PML, but whose CSF PCR for JCV DNA is repeatedly negative, a brain biopsy is appropriate regardless of background immunosuppression. In the patient with rheumatologic disease, for example, suspicion of PML should be heightened if there is neurologic deterioration in the face of escalating antiinflammatory or immunosuppressive therapy for immune-driven inflammatory disease. Diagnostic urgency is particularly warranted in those disorders where the possibility for immune reconstitution is highest (ie, those receiving immunosuppressive regimens).
PML MANAGEMENT DEPENDS ON CLINICAL SETTING
Management of PML starts with risk stratification to identify those patients most prone to developing PML based on their immune status; the presence of autoimmune disease; the subtype of disease (in the case of SLE); and the nature, intensity, and duration of their immunosuppression. If a high-risk patient develops signs and symptoms of PML, the diagnosis should be anticipated and serious consideration given to withholding immunosuppressive therapies while the patient is being worked up.
Accelerating immune reconstitution
Once a diagnosis of PML is confirmed, immune reconstitution should be accelerated whenever possible. This can include temporary or permanent withdrawal of immunosuppressive therapy and initiation of plasmapheresis. Evidence supports continuing plasma exchange until natalizumab serum drug levels decline to less than 1 μg/mL to achieve desaturation of the alpha-4 integrin receptor.6 Typically, desaturation of the targeted integrin receptor occurs after five plasmapheresis sessions.
Immune reconstitution may also precipitate a syndrome known as the immune reconstitution inflammatory syndrome (IRIS), characterized by enlargement and contrast enhancement of PML lesions, appearance of new brain lesions, and worsening of neurologic deficits. The infiltration of the brain with inflammatory multinucleated cells and lymphocytes following abrupt immune reconstitution requires treatment. Opinion suggests that judicious use of corticosteroids may control the immune response in the brain in patients with PML-IRIS,7,8 although further studies are needed.
Involving the patient in treatment decisions
Because risk tolerance varies considerably among individuals, patients should be informed of the risks of PML on the basis of their disease and the agents used to treat it. They should also be given information about the effects of individual treatments on the course of their disease, and they should be encouraged to participate in the selection of therapy.
SUMMARY
The approach to PML in the biologic era starts with an increased awareness of the disease followed by recognition of vulnerable populations and factors that contribute to the development of PML, such as biologic and nonbiologic immunosuppressive therapy. Optimal management includes a low threshold for investigating neurologic signs and symptoms and new-onset signs and symptoms in vulnerable populations, the use of MRI to detect typical PML brain lesions and other atypical brain features (ie, cerebellar atrophy), lumbar puncture and spinal CSF analysis to detect JCV DNA, and timely neurologic consultation for further evaluation. Much still needs to be learned about PML and the risks imparted by background diseases and individual drugs used in rheumatologic, neurologic, and oncologic disease.
Our remarkable progress in understanding progressive multifocal leukoencephalopathy (PML) since its discovery more than 50 years ago has evolved in three stages, concurrent with the changing epidemiology of PML: the pre–human immunodeficiency virus (HIV) era; the HIV era, with highly active antiretroviral therapy (HAART) bringing further change; and the biologic therapy era.
Before the appearance of HIV, PML developed mostly in patients who had lymphoma, other malignancies, and rare forms of immunosuppression. The development of HIV changed the nature of PML, with more than 75% of cases now reported in HIV-infected patients. Within the HIV population, the epidemiology and prognosis of PML have undergone additional changes since the late 1990s. The introduction of HAART transformed PML from an almost uniformly fatal and inexorably progressive disease to one in which long-term survival is expected, particularly in the setting of robust immune reconstitution.1
The third and most recent stage in the evolution of PML and our understanding of it has coincided with the introduction and use of increasingly potent immunosuppressive regimens and novel biologic immunologic therapies that target various aspects of the integrated immune response. These agents are being applied not only in the field of autoimmune and autoinflammatory disease but also in transplantation and oncology.
Collectively, vulnerable populations (ie, patients with lymphoreticular malignancies and autoinflammatory diseases) are now being subjected to therapies that singly or in combination have unknown effects on the immune system. As a byproduct, practitioners who were only vaguely aware of PML in the past now must consider PML in their differential diagnosis, develop a rational plan for evaluating such patients, and recognize when referral to a specialist is indicated. Recent descriptions of atypical forms of PML2,3 add to the challenge for clinicians, as do reports of cases of PML in patients with minimal immunosuppression, in the absence of immunosuppressive therapy, and in patients who appear to have “normal” immune systems but in fact have diseases such as sarcoidosis.4 Rare cases are also being reported in patients with advanced liver disease.4
This article offers recommendations for enhanced awareness of PML, suggestions for improved evaluation of predisposed patients, and a summary of currently accepted treatment strategies.
WHEN TO SUSPECT PML
Patients being treated with immunosuppressive biologic agents represent a significant group that is predisposed to PML. At one time, focal neurologic deficits were required to consider the possibility of PML, but cognitive/behavioral abnormalities rather than focal neurologic findings are often the presenting sign in individuals treated with immune-modulating biologic agents. This phenomenon is most strikingly observed in recipients of natalizumab. Any central nervous system (CNS) dysfunction in a patient taking an immunosuppressive biologic agent should arouse suspicion for PML.
Peripheral neuropathy is not caused by PML but can coexist with it. Accordingly, in patients with rheumatologic disease who are receiving immune modifiers, neuromuscular symptoms in the absence of brain abnormalities on magnetic resonance imaging (MRI) argue against consideration of PML but do not rule it out—especially in patients with connective tissue diseases.
Systemic lupus erythematosus (SLE) represents a special challenge for several reasons. First, SLE appears to be a predisposing factor among other connective tissue diseases.5 In addition, SLE is associated with a variety of CNS complications, including a spectrum of focal and diffuse signs and symptoms that can mimic PML and lead to underdiagnosis.
Underrecognition is a risk in the HIV population as well, where cognitive impairment is common. Irrespective of immune or virologic status, 57% of HIV patients demonstrate impairment on neuropsychiatric testing. Often, mild to moderate cognitive impairment in HIV is attributed to HIV encephalopathy with no further workup, resulting in a missed or late diagnosis of PML.
IMAGING CONSIDERATIONS
In the rheumatologic disease population, especially those with SLE, and the HIV population, neuroimaging is indicated in any patient who presents with cognitive impairment. Typical radiographic characteristics of PML on MRI are subcortical white matter hyperintense areas on T2-weighted images and fluid-attenuated inversion recovery. T1-weighted images will reveal hypointense lesions that usually do not enhance, but may do so in fewer than 10% of patients with PML. Typically, no mass effect is seen.
In addition to rare faint gadolinium enhancement of lesions, other lesion characteristics may depart from the classic picture—for example, white matter and gray matter involvement, and monofocal instead of multifocal lesions. In HIV-positive patients, MRI can demonstrate diffuse cerebellar atrophy and subtle white matter abnormalities within the cerebellum.
Unfortunately, nonspecific white matter lesions occur in HIV infection as well as connective tissue diseases, compromising diagnostic specificity of a single imaging study. Nevertheless, progression of clinical signs and symptoms and progressive MRI changes should prompt a more vigorous diagnostic evaluation for PML. Alternatively, a normal MRI in a patient in whom PML is suspected has strong negative predictive value. In either situation, baseline neuroimaging is not recommended.
DIAGNOSIS AND REFERRAL
A neurology consult is advised when a patient has a predisposing condition for PML or suspicious neurologic signs or symptoms, whether focal or diffuse, and in whom an MRI demonstrates white matter changes.
Evaluation for JC virus DNA
When the neurology consult has been scheduled, and before the actual visit to the neurologist, a cerebrospinal fluid (CSF) sample should be obtained and evaluated for JC virus (JCV) DNA using a highly sensitive polymerase chain reaction (PCR) assay. Lumbar puncture in the setting of possible PML is critical to exclude the presence of other opportunistic infections.
The importance of using ultrasensitive PCR assays for diagnosing PML cannot be overstated, as falsely negative CSF PCR has been observed for JCV DNA despite high levels of JCV DNA in spinal fluid when utilizing less sensitive assays. The most sensitive commercial assays can detect as few as 50 copies of JCV DNA per mL of CSF fluid.
The risk of PML imparted by biologic agents other than natalizumab and nonbiologic immunosuppressive agents has been difficult to quantify, but no immune-modifying drug or combination of drugs appears entirely free of risk. Any patient who has had significant or prolonged immunosuppression should be considered vulnerable, and any patient with suspicion of PML based on unexplained neurologic symptoms warrants CSF examination for JCV DNA.
Brain biopsy
In patients with progressive clinical and MRI findings that suggest PML, but whose CSF PCR for JCV DNA is repeatedly negative, a brain biopsy is appropriate regardless of background immunosuppression. In the patient with rheumatologic disease, for example, suspicion of PML should be heightened if there is neurologic deterioration in the face of escalating antiinflammatory or immunosuppressive therapy for immune-driven inflammatory disease. Diagnostic urgency is particularly warranted in those disorders where the possibility for immune reconstitution is highest (ie, those receiving immunosuppressive regimens).
PML MANAGEMENT DEPENDS ON CLINICAL SETTING
Management of PML starts with risk stratification to identify those patients most prone to developing PML based on their immune status; the presence of autoimmune disease; the subtype of disease (in the case of SLE); and the nature, intensity, and duration of their immunosuppression. If a high-risk patient develops signs and symptoms of PML, the diagnosis should be anticipated and serious consideration given to withholding immunosuppressive therapies while the patient is being worked up.
Accelerating immune reconstitution
Once a diagnosis of PML is confirmed, immune reconstitution should be accelerated whenever possible. This can include temporary or permanent withdrawal of immunosuppressive therapy and initiation of plasmapheresis. Evidence supports continuing plasma exchange until natalizumab serum drug levels decline to less than 1 μg/mL to achieve desaturation of the alpha-4 integrin receptor.6 Typically, desaturation of the targeted integrin receptor occurs after five plasmapheresis sessions.
Immune reconstitution may also precipitate a syndrome known as the immune reconstitution inflammatory syndrome (IRIS), characterized by enlargement and contrast enhancement of PML lesions, appearance of new brain lesions, and worsening of neurologic deficits. The infiltration of the brain with inflammatory multinucleated cells and lymphocytes following abrupt immune reconstitution requires treatment. Opinion suggests that judicious use of corticosteroids may control the immune response in the brain in patients with PML-IRIS,7,8 although further studies are needed.
Involving the patient in treatment decisions
Because risk tolerance varies considerably among individuals, patients should be informed of the risks of PML on the basis of their disease and the agents used to treat it. They should also be given information about the effects of individual treatments on the course of their disease, and they should be encouraged to participate in the selection of therapy.
SUMMARY
The approach to PML in the biologic era starts with an increased awareness of the disease followed by recognition of vulnerable populations and factors that contribute to the development of PML, such as biologic and nonbiologic immunosuppressive therapy. Optimal management includes a low threshold for investigating neurologic signs and symptoms and new-onset signs and symptoms in vulnerable populations, the use of MRI to detect typical PML brain lesions and other atypical brain features (ie, cerebellar atrophy), lumbar puncture and spinal CSF analysis to detect JCV DNA, and timely neurologic consultation for further evaluation. Much still needs to be learned about PML and the risks imparted by background diseases and individual drugs used in rheumatologic, neurologic, and oncologic disease.
- Brew BJ, Davies NW, Cinque P, Clifford DB, Nath A. Progressive multifocal leukoencephalopathy and other forms of JC virus disease. Nat Rev Neurol 2010; 6:667–679.
- Kedar S, Berger JR. The changing landscape of progressive multifocal leukoencephalopathy. Curr Infect Dis Rep 2011; 13:380–386.
- Tan CS, Koralnik IJ. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol 2010; 9:425–437.
- Gheuens S, Pierone G, Peeters P, Koralnik IJ. Progressive multifocal leukoencephalopathy in individuals with minimal or occult immunosuppression. J Neurol Neurosurg Psychiatry 2010; 81:247–254.
- Molloy ES, Calabrese LH. Progressive multifocal leukoencephalopathy: a national estimate of frequency in systemic lupus erythematosus and other rheumatic diseases. Arthritis Rheum 2009; 60:3761–3765.
- Khatri BO, Man S, Giovannoni G, et al. Effect of plasma exchange in accelerating natalizumab clearance and restoring leukocyte function. Neurology 2009; 72:402–409.
- Clifford DB, De Luca A, Simpson DM, Arendt G, Giovannoni G, Nath A. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol 2010; 9:438–446.
- Johnson T, Nath A. Immune reconstitution inflammatory syndrome and the central nervous system. Curr Opin Neurol 2011; 24:284–290.
- Brew BJ, Davies NW, Cinque P, Clifford DB, Nath A. Progressive multifocal leukoencephalopathy and other forms of JC virus disease. Nat Rev Neurol 2010; 6:667–679.
- Kedar S, Berger JR. The changing landscape of progressive multifocal leukoencephalopathy. Curr Infect Dis Rep 2011; 13:380–386.
- Tan CS, Koralnik IJ. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol 2010; 9:425–437.
- Gheuens S, Pierone G, Peeters P, Koralnik IJ. Progressive multifocal leukoencephalopathy in individuals with minimal or occult immunosuppression. J Neurol Neurosurg Psychiatry 2010; 81:247–254.
- Molloy ES, Calabrese LH. Progressive multifocal leukoencephalopathy: a national estimate of frequency in systemic lupus erythematosus and other rheumatic diseases. Arthritis Rheum 2009; 60:3761–3765.
- Khatri BO, Man S, Giovannoni G, et al. Effect of plasma exchange in accelerating natalizumab clearance and restoring leukocyte function. Neurology 2009; 72:402–409.
- Clifford DB, De Luca A, Simpson DM, Arendt G, Giovannoni G, Nath A. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases. Lancet Neurol 2010; 9:438–446.
- Johnson T, Nath A. Immune reconstitution inflammatory syndrome and the central nervous system. Curr Opin Neurol 2011; 24:284–290.