User login
Venetoclax approved to treat CLL patients regardless of genotype
The approval of Bcl-2 inhibitor venetoclax was expanded by the US Food and Drug Administration in June 2018 to include the treatment of patients with chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL), regardless of their genotype, who have received at least 1 prior therapy.1 It was previously approved in 2016 for the treatment of patients who had a chromosome 17p deletion, which leads to loss of the tumor-suppressor gene TP53.
Approval was based on the positive results of the phase 3, randomized, multicenter, open-label MURANO trial in which 389 patients were randomized 1:1 to receive a combination of venetoclax and the CD20-targeting monoclonal antibody rituximab (venetoclax–rituximab) or bendamustine in combination with rituximab (bendamustine–rituximab).
Eligible patients were 18 years of age or older, had been diagnosed with relapsed/refractory CLL that required treatment, had received 1-3 prior therapies (including at least 1 chemotherapy regimen), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (on a 5-point scale, with 5 indicating the greatest level of disability), and had adequate bone marrow, renal, and hepatic function.
Patients who had received prior bendamustine treatment were eligible for the trial provided they had experienced a duration of response of 24 months or longer. However, patients with transformed CLL, central nervous system involvement, prior treatment with allogeneic or autologous stem cell transplant, major organ dysfunction, other active malignancy, or who were pregnant or breastfeeding, were excluded from the study.
Patients in the venetoclax arm received a 5-week ramp-up schedule, followed by a dose of 400 mg once daily for 24 months. Rituximab treatment started at the end of the venetoclax ramp-up period and was administered at a dose of 375 mg/m2 intravenously on cycle 1 day 1 and 500 mg/m2 on day 1 of cycles 2-6. In the control arm, patients received 6 cycles with the same rituxima
The primary endpoint was progression-free survival (PFS), as assessed by an independent review committee over a median follow-up of 23 months. Median PFS was significantly improved in the venetoclax arm (not yet reached versus 18.1 months in the bendamustine arm [HR, 0.19; P < .001]). In addition, objective response rate (ORR) and event-free survival (EFS) also favored the venetoclax arm; ORR was 92% compared with 72%, respectively, and 2-year EFS was 84.9% compared with 34.8%. There was also a trend toward improved 24-month overall survival (OS) rate (91.9% vs 86.6%), however this did not achieve statistical significance, nor did median OS.
The most common adverse events (AEs) in patients treated with venetoclax were neutropenia, diarrhea, upper-respiratory tract infection, fatigue, cough, and nausea. Grade 3/4 neutropenia occurred in 64% of patients, and serious AEs in 46% of patients. Serious infections occurred in 21% of patients, most commonly pneumonia. Ten deaths in the venetoclax arm were attributed to treatment, compared with 11 deaths in the bendamustine arm.2
The prescribing information details warnings and precautions relating to the risk of tumor lysis syndrome, which is increased in patients with higher tumor burden, reduced renal function, or in receipt of strong or moderate CYP3A inhibitors or P-gp inhibitors during the ramp-up stage. Patients should receive appropriate preventive strategies, including hydration and antihyperuricemics, blood chemistry should be monitored and abnormalities managed promptly, and dosing should be interrupted or adjusted as necessary.
Other warnings relate to neutropenia (complete blood counts should be monitored throughout treatment and venetoclax treatment interrupted or dose reduced for severe neutropenia, alongside possible use of supportive measures), immunization (live vaccines should not be administered before or during treatment or after treatment until B-cell recovery, and patients should be advised of the potentially reduced efficacy of vaccines), and embryofetal toxicity (patients should be advised of the risks and the need for effective contraception during and after treatment). Venetoclax is marketed as Venclexta by Genentech.3
1. US Food and Drug Administration website. FDA approves venetoclax for CLL or SLL, with or without 17p deletion, after one prior therapy. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm610308.htm. Last updated June 8, 2018. Accessed July 29, 2018.
2. Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378:1107-1120.
3. Venclexta (venetoclax tablets) for oral use. Prescribing information. Genentech USA, Inc. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/208573s000lbl.pdf. Last updated June 2018. Accessed July 29, 2018.
The approval of Bcl-2 inhibitor venetoclax was expanded by the US Food and Drug Administration in June 2018 to include the treatment of patients with chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL), regardless of their genotype, who have received at least 1 prior therapy.1 It was previously approved in 2016 for the treatment of patients who had a chromosome 17p deletion, which leads to loss of the tumor-suppressor gene TP53.
Approval was based on the positive results of the phase 3, randomized, multicenter, open-label MURANO trial in which 389 patients were randomized 1:1 to receive a combination of venetoclax and the CD20-targeting monoclonal antibody rituximab (venetoclax–rituximab) or bendamustine in combination with rituximab (bendamustine–rituximab).
Eligible patients were 18 years of age or older, had been diagnosed with relapsed/refractory CLL that required treatment, had received 1-3 prior therapies (including at least 1 chemotherapy regimen), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (on a 5-point scale, with 5 indicating the greatest level of disability), and had adequate bone marrow, renal, and hepatic function.
Patients who had received prior bendamustine treatment were eligible for the trial provided they had experienced a duration of response of 24 months or longer. However, patients with transformed CLL, central nervous system involvement, prior treatment with allogeneic or autologous stem cell transplant, major organ dysfunction, other active malignancy, or who were pregnant or breastfeeding, were excluded from the study.
Patients in the venetoclax arm received a 5-week ramp-up schedule, followed by a dose of 400 mg once daily for 24 months. Rituximab treatment started at the end of the venetoclax ramp-up period and was administered at a dose of 375 mg/m2 intravenously on cycle 1 day 1 and 500 mg/m2 on day 1 of cycles 2-6. In the control arm, patients received 6 cycles with the same rituxima
The primary endpoint was progression-free survival (PFS), as assessed by an independent review committee over a median follow-up of 23 months. Median PFS was significantly improved in the venetoclax arm (not yet reached versus 18.1 months in the bendamustine arm [HR, 0.19; P < .001]). In addition, objective response rate (ORR) and event-free survival (EFS) also favored the venetoclax arm; ORR was 92% compared with 72%, respectively, and 2-year EFS was 84.9% compared with 34.8%. There was also a trend toward improved 24-month overall survival (OS) rate (91.9% vs 86.6%), however this did not achieve statistical significance, nor did median OS.
The most common adverse events (AEs) in patients treated with venetoclax were neutropenia, diarrhea, upper-respiratory tract infection, fatigue, cough, and nausea. Grade 3/4 neutropenia occurred in 64% of patients, and serious AEs in 46% of patients. Serious infections occurred in 21% of patients, most commonly pneumonia. Ten deaths in the venetoclax arm were attributed to treatment, compared with 11 deaths in the bendamustine arm.2
The prescribing information details warnings and precautions relating to the risk of tumor lysis syndrome, which is increased in patients with higher tumor burden, reduced renal function, or in receipt of strong or moderate CYP3A inhibitors or P-gp inhibitors during the ramp-up stage. Patients should receive appropriate preventive strategies, including hydration and antihyperuricemics, blood chemistry should be monitored and abnormalities managed promptly, and dosing should be interrupted or adjusted as necessary.
Other warnings relate to neutropenia (complete blood counts should be monitored throughout treatment and venetoclax treatment interrupted or dose reduced for severe neutropenia, alongside possible use of supportive measures), immunization (live vaccines should not be administered before or during treatment or after treatment until B-cell recovery, and patients should be advised of the potentially reduced efficacy of vaccines), and embryofetal toxicity (patients should be advised of the risks and the need for effective contraception during and after treatment). Venetoclax is marketed as Venclexta by Genentech.3
The approval of Bcl-2 inhibitor venetoclax was expanded by the US Food and Drug Administration in June 2018 to include the treatment of patients with chronic lymphocytic leukemia (CLL) or small lymphocytic leukemia (SLL), regardless of their genotype, who have received at least 1 prior therapy.1 It was previously approved in 2016 for the treatment of patients who had a chromosome 17p deletion, which leads to loss of the tumor-suppressor gene TP53.
Approval was based on the positive results of the phase 3, randomized, multicenter, open-label MURANO trial in which 389 patients were randomized 1:1 to receive a combination of venetoclax and the CD20-targeting monoclonal antibody rituximab (venetoclax–rituximab) or bendamustine in combination with rituximab (bendamustine–rituximab).
Eligible patients were 18 years of age or older, had been diagnosed with relapsed/refractory CLL that required treatment, had received 1-3 prior therapies (including at least 1 chemotherapy regimen), had an Eastern Cooperative Oncology Group performance status of 0 or 1 (on a 5-point scale, with 5 indicating the greatest level of disability), and had adequate bone marrow, renal, and hepatic function.
Patients who had received prior bendamustine treatment were eligible for the trial provided they had experienced a duration of response of 24 months or longer. However, patients with transformed CLL, central nervous system involvement, prior treatment with allogeneic or autologous stem cell transplant, major organ dysfunction, other active malignancy, or who were pregnant or breastfeeding, were excluded from the study.
Patients in the venetoclax arm received a 5-week ramp-up schedule, followed by a dose of 400 mg once daily for 24 months. Rituximab treatment started at the end of the venetoclax ramp-up period and was administered at a dose of 375 mg/m2 intravenously on cycle 1 day 1 and 500 mg/m2 on day 1 of cycles 2-6. In the control arm, patients received 6 cycles with the same rituxima
The primary endpoint was progression-free survival (PFS), as assessed by an independent review committee over a median follow-up of 23 months. Median PFS was significantly improved in the venetoclax arm (not yet reached versus 18.1 months in the bendamustine arm [HR, 0.19; P < .001]). In addition, objective response rate (ORR) and event-free survival (EFS) also favored the venetoclax arm; ORR was 92% compared with 72%, respectively, and 2-year EFS was 84.9% compared with 34.8%. There was also a trend toward improved 24-month overall survival (OS) rate (91.9% vs 86.6%), however this did not achieve statistical significance, nor did median OS.
The most common adverse events (AEs) in patients treated with venetoclax were neutropenia, diarrhea, upper-respiratory tract infection, fatigue, cough, and nausea. Grade 3/4 neutropenia occurred in 64% of patients, and serious AEs in 46% of patients. Serious infections occurred in 21% of patients, most commonly pneumonia. Ten deaths in the venetoclax arm were attributed to treatment, compared with 11 deaths in the bendamustine arm.2
The prescribing information details warnings and precautions relating to the risk of tumor lysis syndrome, which is increased in patients with higher tumor burden, reduced renal function, or in receipt of strong or moderate CYP3A inhibitors or P-gp inhibitors during the ramp-up stage. Patients should receive appropriate preventive strategies, including hydration and antihyperuricemics, blood chemistry should be monitored and abnormalities managed promptly, and dosing should be interrupted or adjusted as necessary.
Other warnings relate to neutropenia (complete blood counts should be monitored throughout treatment and venetoclax treatment interrupted or dose reduced for severe neutropenia, alongside possible use of supportive measures), immunization (live vaccines should not be administered before or during treatment or after treatment until B-cell recovery, and patients should be advised of the potentially reduced efficacy of vaccines), and embryofetal toxicity (patients should be advised of the risks and the need for effective contraception during and after treatment). Venetoclax is marketed as Venclexta by Genentech.3
1. US Food and Drug Administration website. FDA approves venetoclax for CLL or SLL, with or without 17p deletion, after one prior therapy. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm610308.htm. Last updated June 8, 2018. Accessed July 29, 2018.
2. Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378:1107-1120.
3. Venclexta (venetoclax tablets) for oral use. Prescribing information. Genentech USA, Inc. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/208573s000lbl.pdf. Last updated June 2018. Accessed July 29, 2018.
1. US Food and Drug Administration website. FDA approves venetoclax for CLL or SLL, with or without 17p deletion, after one prior therapy. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm610308.htm. Last updated June 8, 2018. Accessed July 29, 2018.
2. Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378:1107-1120.
3. Venclexta (venetoclax tablets) for oral use. Prescribing information. Genentech USA, Inc. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/208573s000lbl.pdf. Last updated June 2018. Accessed July 29, 2018.
Nivolumab and ipilimumab combination promises new standard of care for advanced RCC
In April 2018, the US Food and Drug Administration expanded the approval of the combination of nivolumab and ipilimumab into a new indication, following a previous approval in patients with metastatic melanoma. The double immune checkpoint inhibitor combination was approved on the basis of the phase 3 CheckMate-214 study for the treatment of patients with intermediate- or poor-risk, previously untreated advanced renal cell carcinoma (RCC).1
Nivolumab monotherapy is already approved in the second-line setting for the treatment of advanced RCC, and the demonstration of significantly improved overall survival (OS) in this study suggests that the combination should supplant sunitinib in the front-line setting in the treatment of this type of cancer.
A total of 1,096 patients at 175 sites in 28 countries were randomized 1:1 to receive nivolumab (3 mg/kg) and ipilimumab (1 mg/kg) intravenously every 3 weeks for 4 doses in an induction phase, followed by nivolumab monotherapy (3 mg/kg) every 2 weeks in a maintenance phase or sunitinib (50 mg) orally daily for 4 weeks of each 6-week cycle.
Eligible patients were 18 years or older, had previously untreated advanced RCC with a clear-cell component, had measurable disease according to Response Evaluation Criteria in Solid Tumors (version 1.1), and had a Karnofsky performance status of at least 70 (on a scale from 0 to 100, with lower scores indicating greater disability). Patients with central nervous system metastases or autoimmune disease who were being treated with glucocorticoids and immunosuppressants were excluded from the study.
Around three-quarters of patients with advanced RCC have intermediate- or poor-risk disease and experience worse outcomes than patients with favorable-risk disease. Patients in CheckMate-214 were stratified according to International Metastatic Renal Cell Carcinoma Database Consortium risk score as favorable (score of 0), intermediate (score of 1 or 2) or poor risk (score of 3-6), according to the number of risk factors present.
Risk factors included a Karnofsky performance score of 70, time from initial diagnosis to randomization of <1 year, a hemoglobin level below the lower limit of normal, a corrected serum calcium concentration of >10 mg/dL, or an absolute neutrophil count or platelet count above the upper limit of normal. Patients were also stratified according to geographic region (United States versus Canada and Europe versus the rest of the world).
The coprimary endpoints were objective response rate (ORR), progression-free survival (PFS), and OS in a subset of 847 intermediate- and poor-risk patients. Over a median follow-up of 25.2 months, there was a statistically significant improvement in OS and ORR in patients treated with nivolumab and ipilimumab (mPFS not reached; ORR, 41.6%), compared with sunitinib (OS, 25.9 months; ORR, 26.5%), with P <.001 for both. The immunotherapy combination was favored across subgroups.
The most common adverse events (AEs) in patients treated with nivolumab and ipilimumab included fatigue, rash, diarrhea, musculoskeletal pain, pruritus, nausea, cough, pyrexia, arthralgia, and decreased appetite. The combination was associated with fewer grade 3/4 AEs (63% vs 46% for sunitinib), but a higher rate of treatment discontinuations because of AEs (31% vs 21%, respectively). There were 8 deaths in the combination arm, and 4 in the sunitinib arm that were reported to be treatment related.2
The warnings and precautions related to nivolumab–ipilimumab combination therapy outlined in the prescribing information include mostly immune-mediated AEs, such as immune-mediated pneumonitis, colitis, hepatitis, endocrinopathies, nephritis and renal dysfunction, skin adverse reactions, and encephalitis. There are also warnings relating to the risk of infusion reactions and the potential for embryofetal toxicity.
Patients should be monitored for hyperglycemia and for changes in liver, thyroid, renal, and neurologic function. Treatment with nivolumab and ipilimumab should be withheld for moderate and permanently discontinued for severe or life-threatening immune-mediated pneumonitis, colitis, and hepatitis, as well as transaminase or total bilirubin elevation. It should also be withheld for moderate or severe hypophysitis and serum creatinine elevation, moderate adrenal insufficiency and severe hyperglycemia, and permanently discontinued for life-threatening hypophysitis and serum creatinine elevation, severe or life-threatening adrenal insufficiency, and life-threatening hyperglycemia.
New-onset moderate to severe neurologic signs or symptoms warrant treatment being withheld, and immune-mediated encephalitis should lead to treatment discontinuation. For mild or moderate infusion reactions, the infusion rate can be slowed or interrupted, and infusions should be discontinued in the event of severe or life-threatening infusion reactions. Patients should be advised of the potential for fetal harm and the need for effective contraception during and after treatment. Ipilimumab and nivolumab are marketed as Yervoy and Opdivo, respectively, by Bristol-Myers Squibb.3,4
1. US Food and Drug Administration website. FDA approves nivolumab plus ipilimumab combination for intermediate or poor-risk advanced renal cell carcinoma. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm604685.htm. Last updated April 16, 2018. Accessed July 25, 2018.
2. Motzer RJ, Tannir NM, McDermott O, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378:1277-1290.
3. Opdivo (nivolumab) injection, for intravenous use. Prescribing information. Bristol-Myers Squibb. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125554s058lbl.pdf. Revised April 2018.
4. Yervoy (ipilimumab) injection, for intravenous use. Prescribing information. Bristol-Myers Squibb. July 2018. https://packageinserts.bms.com/pi/pi_yervoy.pdf. Accessed September, 2018.
In April 2018, the US Food and Drug Administration expanded the approval of the combination of nivolumab and ipilimumab into a new indication, following a previous approval in patients with metastatic melanoma. The double immune checkpoint inhibitor combination was approved on the basis of the phase 3 CheckMate-214 study for the treatment of patients with intermediate- or poor-risk, previously untreated advanced renal cell carcinoma (RCC).1
Nivolumab monotherapy is already approved in the second-line setting for the treatment of advanced RCC, and the demonstration of significantly improved overall survival (OS) in this study suggests that the combination should supplant sunitinib in the front-line setting in the treatment of this type of cancer.
A total of 1,096 patients at 175 sites in 28 countries were randomized 1:1 to receive nivolumab (3 mg/kg) and ipilimumab (1 mg/kg) intravenously every 3 weeks for 4 doses in an induction phase, followed by nivolumab monotherapy (3 mg/kg) every 2 weeks in a maintenance phase or sunitinib (50 mg) orally daily for 4 weeks of each 6-week cycle.
Eligible patients were 18 years or older, had previously untreated advanced RCC with a clear-cell component, had measurable disease according to Response Evaluation Criteria in Solid Tumors (version 1.1), and had a Karnofsky performance status of at least 70 (on a scale from 0 to 100, with lower scores indicating greater disability). Patients with central nervous system metastases or autoimmune disease who were being treated with glucocorticoids and immunosuppressants were excluded from the study.
Around three-quarters of patients with advanced RCC have intermediate- or poor-risk disease and experience worse outcomes than patients with favorable-risk disease. Patients in CheckMate-214 were stratified according to International Metastatic Renal Cell Carcinoma Database Consortium risk score as favorable (score of 0), intermediate (score of 1 or 2) or poor risk (score of 3-6), according to the number of risk factors present.
Risk factors included a Karnofsky performance score of 70, time from initial diagnosis to randomization of <1 year, a hemoglobin level below the lower limit of normal, a corrected serum calcium concentration of >10 mg/dL, or an absolute neutrophil count or platelet count above the upper limit of normal. Patients were also stratified according to geographic region (United States versus Canada and Europe versus the rest of the world).
The coprimary endpoints were objective response rate (ORR), progression-free survival (PFS), and OS in a subset of 847 intermediate- and poor-risk patients. Over a median follow-up of 25.2 months, there was a statistically significant improvement in OS and ORR in patients treated with nivolumab and ipilimumab (mPFS not reached; ORR, 41.6%), compared with sunitinib (OS, 25.9 months; ORR, 26.5%), with P <.001 for both. The immunotherapy combination was favored across subgroups.
The most common adverse events (AEs) in patients treated with nivolumab and ipilimumab included fatigue, rash, diarrhea, musculoskeletal pain, pruritus, nausea, cough, pyrexia, arthralgia, and decreased appetite. The combination was associated with fewer grade 3/4 AEs (63% vs 46% for sunitinib), but a higher rate of treatment discontinuations because of AEs (31% vs 21%, respectively). There were 8 deaths in the combination arm, and 4 in the sunitinib arm that were reported to be treatment related.2
The warnings and precautions related to nivolumab–ipilimumab combination therapy outlined in the prescribing information include mostly immune-mediated AEs, such as immune-mediated pneumonitis, colitis, hepatitis, endocrinopathies, nephritis and renal dysfunction, skin adverse reactions, and encephalitis. There are also warnings relating to the risk of infusion reactions and the potential for embryofetal toxicity.
Patients should be monitored for hyperglycemia and for changes in liver, thyroid, renal, and neurologic function. Treatment with nivolumab and ipilimumab should be withheld for moderate and permanently discontinued for severe or life-threatening immune-mediated pneumonitis, colitis, and hepatitis, as well as transaminase or total bilirubin elevation. It should also be withheld for moderate or severe hypophysitis and serum creatinine elevation, moderate adrenal insufficiency and severe hyperglycemia, and permanently discontinued for life-threatening hypophysitis and serum creatinine elevation, severe or life-threatening adrenal insufficiency, and life-threatening hyperglycemia.
New-onset moderate to severe neurologic signs or symptoms warrant treatment being withheld, and immune-mediated encephalitis should lead to treatment discontinuation. For mild or moderate infusion reactions, the infusion rate can be slowed or interrupted, and infusions should be discontinued in the event of severe or life-threatening infusion reactions. Patients should be advised of the potential for fetal harm and the need for effective contraception during and after treatment. Ipilimumab and nivolumab are marketed as Yervoy and Opdivo, respectively, by Bristol-Myers Squibb.3,4
In April 2018, the US Food and Drug Administration expanded the approval of the combination of nivolumab and ipilimumab into a new indication, following a previous approval in patients with metastatic melanoma. The double immune checkpoint inhibitor combination was approved on the basis of the phase 3 CheckMate-214 study for the treatment of patients with intermediate- or poor-risk, previously untreated advanced renal cell carcinoma (RCC).1
Nivolumab monotherapy is already approved in the second-line setting for the treatment of advanced RCC, and the demonstration of significantly improved overall survival (OS) in this study suggests that the combination should supplant sunitinib in the front-line setting in the treatment of this type of cancer.
A total of 1,096 patients at 175 sites in 28 countries were randomized 1:1 to receive nivolumab (3 mg/kg) and ipilimumab (1 mg/kg) intravenously every 3 weeks for 4 doses in an induction phase, followed by nivolumab monotherapy (3 mg/kg) every 2 weeks in a maintenance phase or sunitinib (50 mg) orally daily for 4 weeks of each 6-week cycle.
Eligible patients were 18 years or older, had previously untreated advanced RCC with a clear-cell component, had measurable disease according to Response Evaluation Criteria in Solid Tumors (version 1.1), and had a Karnofsky performance status of at least 70 (on a scale from 0 to 100, with lower scores indicating greater disability). Patients with central nervous system metastases or autoimmune disease who were being treated with glucocorticoids and immunosuppressants were excluded from the study.
Around three-quarters of patients with advanced RCC have intermediate- or poor-risk disease and experience worse outcomes than patients with favorable-risk disease. Patients in CheckMate-214 were stratified according to International Metastatic Renal Cell Carcinoma Database Consortium risk score as favorable (score of 0), intermediate (score of 1 or 2) or poor risk (score of 3-6), according to the number of risk factors present.
Risk factors included a Karnofsky performance score of 70, time from initial diagnosis to randomization of <1 year, a hemoglobin level below the lower limit of normal, a corrected serum calcium concentration of >10 mg/dL, or an absolute neutrophil count or platelet count above the upper limit of normal. Patients were also stratified according to geographic region (United States versus Canada and Europe versus the rest of the world).
The coprimary endpoints were objective response rate (ORR), progression-free survival (PFS), and OS in a subset of 847 intermediate- and poor-risk patients. Over a median follow-up of 25.2 months, there was a statistically significant improvement in OS and ORR in patients treated with nivolumab and ipilimumab (mPFS not reached; ORR, 41.6%), compared with sunitinib (OS, 25.9 months; ORR, 26.5%), with P <.001 for both. The immunotherapy combination was favored across subgroups.
The most common adverse events (AEs) in patients treated with nivolumab and ipilimumab included fatigue, rash, diarrhea, musculoskeletal pain, pruritus, nausea, cough, pyrexia, arthralgia, and decreased appetite. The combination was associated with fewer grade 3/4 AEs (63% vs 46% for sunitinib), but a higher rate of treatment discontinuations because of AEs (31% vs 21%, respectively). There were 8 deaths in the combination arm, and 4 in the sunitinib arm that were reported to be treatment related.2
The warnings and precautions related to nivolumab–ipilimumab combination therapy outlined in the prescribing information include mostly immune-mediated AEs, such as immune-mediated pneumonitis, colitis, hepatitis, endocrinopathies, nephritis and renal dysfunction, skin adverse reactions, and encephalitis. There are also warnings relating to the risk of infusion reactions and the potential for embryofetal toxicity.
Patients should be monitored for hyperglycemia and for changes in liver, thyroid, renal, and neurologic function. Treatment with nivolumab and ipilimumab should be withheld for moderate and permanently discontinued for severe or life-threatening immune-mediated pneumonitis, colitis, and hepatitis, as well as transaminase or total bilirubin elevation. It should also be withheld for moderate or severe hypophysitis and serum creatinine elevation, moderate adrenal insufficiency and severe hyperglycemia, and permanently discontinued for life-threatening hypophysitis and serum creatinine elevation, severe or life-threatening adrenal insufficiency, and life-threatening hyperglycemia.
New-onset moderate to severe neurologic signs or symptoms warrant treatment being withheld, and immune-mediated encephalitis should lead to treatment discontinuation. For mild or moderate infusion reactions, the infusion rate can be slowed or interrupted, and infusions should be discontinued in the event of severe or life-threatening infusion reactions. Patients should be advised of the potential for fetal harm and the need for effective contraception during and after treatment. Ipilimumab and nivolumab are marketed as Yervoy and Opdivo, respectively, by Bristol-Myers Squibb.3,4
1. US Food and Drug Administration website. FDA approves nivolumab plus ipilimumab combination for intermediate or poor-risk advanced renal cell carcinoma. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm604685.htm. Last updated April 16, 2018. Accessed July 25, 2018.
2. Motzer RJ, Tannir NM, McDermott O, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378:1277-1290.
3. Opdivo (nivolumab) injection, for intravenous use. Prescribing information. Bristol-Myers Squibb. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125554s058lbl.pdf. Revised April 2018.
4. Yervoy (ipilimumab) injection, for intravenous use. Prescribing information. Bristol-Myers Squibb. July 2018. https://packageinserts.bms.com/pi/pi_yervoy.pdf. Accessed September, 2018.
1. US Food and Drug Administration website. FDA approves nivolumab plus ipilimumab combination for intermediate or poor-risk advanced renal cell carcinoma. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm604685.htm. Last updated April 16, 2018. Accessed July 25, 2018.
2. Motzer RJ, Tannir NM, McDermott O, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378:1277-1290.
3. Opdivo (nivolumab) injection, for intravenous use. Prescribing information. Bristol-Myers Squibb. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125554s058lbl.pdf. Revised April 2018.
4. Yervoy (ipilimumab) injection, for intravenous use. Prescribing information. Bristol-Myers Squibb. July 2018. https://packageinserts.bms.com/pi/pi_yervoy.pdf. Accessed September, 2018.
Neratinib extends adjuvant treatment of patients with HER2-positive breast cancer
The small-molecule tyrosine kinase inhibitor neratinib is now approved for the extended adjuvant treatment of patients with early-stage HER2 [human epidermal growth factor receptor]-positive breast cancer following postoperative trastuzumab. Trastuzumab is a HER2-targeted monoclonal antibody that has become standard of care in combination with chemotherapy for the treatment of this patient population in which it significantly improves survival. However, disease recurrence will occur in about a quarter of trastuzumab-treated patients owing to the development of resistance.
Neratinib may help overcome trastuzumab resistance thanks to its potent inhibition of the downstream phosphorylation of HER2 and other members of the HER family. Its approval was based on the phase 3 ExteNET trial, in which extended adjuvant treatment with neratinib was compared with placebo among 2,840 patients who remained disease free after 1 year of adjuvant trastuzumab.1
The ExteNET trial was performed at 495 centers in Europe, Asia, Australia, New Zealand, and South America. Patients aged 18 years or older (≥20 years in Japan), with stage 1-3 HER2-positive breast cancer, who completed neoadjuvant and adjuvant trastuzumab therapy up to 1 year before randomization were eligible. Patients also had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 (range, 0-5; 0, fully active, and 5, dead), normal organ function, and a left ventricular ejection fraction within normal institutional range. Patients with clinically significant cardiac, gastrointesintal or psychiatric comorbidities and those who were not able to swallow oral medication were excluded from the study.
Patients randomly received oral neratinib 240 mg per day or matching placebo, and randomization was stratified according to HR status (positive or negative), nodal status (0, 1-3, or ≥4) and trastuzumab-adjuvant regimen (sequentially or concurrently with chemotherapy).
The primary outcome was invasive disease-free survival (iDFS). The 2-year iDFS rate was 93.9% for neratinib, compared with 91.6% for placebo (hazard ratio [HR], 0.66; P < .008). Recently, a 5-year analysis of the ExteNET trial showed that after a median follow-up of 5.2 years, the iDFS rates were 90.2% vs 87.7% (HR, 0.73; P = .0083).2
Adverse events
The most common adverse event (AE) was diarrhea, in 95% of patients, 40% of whom had grade 3 diarrhea, leading to dose reduction in 26% of patients and discontinuation in 16.8% of patients. Serious AEs occurred in 7% of patients in the neratinib and 6% of those in the placebo arms. In the 5-year analysis, there was no evidence of increased risk of long-term toxicity or adverse consequences of neratinib-associated diarrhea. Furthermore, the ongoing, open-label phase 2 CONTROL trial suggests that the occurrence and severity of neratinib-associated diarrhea can be effectively controlled with antidiarrheal prophylaxis, with drugs such as loperamide.3
At the January 2017 cut-off, 137 patients treated with neratinib (240 mg/day) for 1 year had also received treatment with loperamide monotherapy, 64 patients had received loperamide and budesonide, and 10 patients had received loperamide and colestipol. The safety data from the loperamide monotherapy arm were compared with the safety data from the ExteNET trial, which was based in a similar population of patients who did not receive antidiarrheal prophylaxis. The incidence of all-grade diarrhea was 77% vs 95%, respectively, for those who received antidiarrheal prophylaxis in the CONTROL trial compared with those in the ExteNET trial who did not, and the repective rates of grade 3 diarrhea were 31% and 40%. The rate of dose reductions and holds owing to diarrhea were also lower among those who received antidiarrheal prophylaxis, but the rate of discontinuation due to diarrhea was higher in the loperamide-treated cohort.
Warnings and precautions
Neratinib is marketed as Nerlynx by Puma Biotechnology Inc. The prescribing information describes warnings and precautions relating to diarrhea, hepatotoxicity, and embryofetal toxicity. Patients should be monitored for diarrhea and treated with antidiarrheals as needed. Severe diarrhea with dehydration should be treated with fluids and electrolytes as needed, treatment should be interrupted and resumed at a reduced dose. For grade 3/4 diarrhea or diarrhea with complicating features (eg, dehydration, fever, neutropenia), stool cultures should be performed to rule out infectious causes.
Total bilirubin, aspartate and alanine aminotransferase, and alkaline phosphatase levels should be measured before starting treatment, every 3 months during treatment, or as clinically indicated. Neratinib can cause fetal harm, so pregnant women should be advised of the risk to the fetus and patients of reproductive potential should be counseled on the need for effective contraception during treatment and for at least 1 month after the last dose.4
1. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17: 367-377.
2. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab- based adjuvant therapy in HER2-positive breast cancer (ExteNET): a 5-year analysis of a randomised, double-blind, placebo- controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1688-1700.
3. Ibrahim E, Tripathy D, Wilkinson M, et al. E£ects of adding budesonide or colestipol to loperamide prophylaxis on neratinib-associated diarrhea in patients (pts) with HER2+ early-stage breast cancer (EBC): The CONTROL trial. Cancer Res. 2017; 77(13 supplement): Abstract CT128.
4. Nerlynx (neratinib) tablets, for oral use. Prescribing information. Puma Biotechnology Inc. https://nerlynx.com/pdf/full-prescribinginformation. pdf. Revised July 2017. Accessed November 20th, 2017.
The small-molecule tyrosine kinase inhibitor neratinib is now approved for the extended adjuvant treatment of patients with early-stage HER2 [human epidermal growth factor receptor]-positive breast cancer following postoperative trastuzumab. Trastuzumab is a HER2-targeted monoclonal antibody that has become standard of care in combination with chemotherapy for the treatment of this patient population in which it significantly improves survival. However, disease recurrence will occur in about a quarter of trastuzumab-treated patients owing to the development of resistance.
Neratinib may help overcome trastuzumab resistance thanks to its potent inhibition of the downstream phosphorylation of HER2 and other members of the HER family. Its approval was based on the phase 3 ExteNET trial, in which extended adjuvant treatment with neratinib was compared with placebo among 2,840 patients who remained disease free after 1 year of adjuvant trastuzumab.1
The ExteNET trial was performed at 495 centers in Europe, Asia, Australia, New Zealand, and South America. Patients aged 18 years or older (≥20 years in Japan), with stage 1-3 HER2-positive breast cancer, who completed neoadjuvant and adjuvant trastuzumab therapy up to 1 year before randomization were eligible. Patients also had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 (range, 0-5; 0, fully active, and 5, dead), normal organ function, and a left ventricular ejection fraction within normal institutional range. Patients with clinically significant cardiac, gastrointesintal or psychiatric comorbidities and those who were not able to swallow oral medication were excluded from the study.
Patients randomly received oral neratinib 240 mg per day or matching placebo, and randomization was stratified according to HR status (positive or negative), nodal status (0, 1-3, or ≥4) and trastuzumab-adjuvant regimen (sequentially or concurrently with chemotherapy).
The primary outcome was invasive disease-free survival (iDFS). The 2-year iDFS rate was 93.9% for neratinib, compared with 91.6% for placebo (hazard ratio [HR], 0.66; P < .008). Recently, a 5-year analysis of the ExteNET trial showed that after a median follow-up of 5.2 years, the iDFS rates were 90.2% vs 87.7% (HR, 0.73; P = .0083).2
Adverse events
The most common adverse event (AE) was diarrhea, in 95% of patients, 40% of whom had grade 3 diarrhea, leading to dose reduction in 26% of patients and discontinuation in 16.8% of patients. Serious AEs occurred in 7% of patients in the neratinib and 6% of those in the placebo arms. In the 5-year analysis, there was no evidence of increased risk of long-term toxicity or adverse consequences of neratinib-associated diarrhea. Furthermore, the ongoing, open-label phase 2 CONTROL trial suggests that the occurrence and severity of neratinib-associated diarrhea can be effectively controlled with antidiarrheal prophylaxis, with drugs such as loperamide.3
At the January 2017 cut-off, 137 patients treated with neratinib (240 mg/day) for 1 year had also received treatment with loperamide monotherapy, 64 patients had received loperamide and budesonide, and 10 patients had received loperamide and colestipol. The safety data from the loperamide monotherapy arm were compared with the safety data from the ExteNET trial, which was based in a similar population of patients who did not receive antidiarrheal prophylaxis. The incidence of all-grade diarrhea was 77% vs 95%, respectively, for those who received antidiarrheal prophylaxis in the CONTROL trial compared with those in the ExteNET trial who did not, and the repective rates of grade 3 diarrhea were 31% and 40%. The rate of dose reductions and holds owing to diarrhea were also lower among those who received antidiarrheal prophylaxis, but the rate of discontinuation due to diarrhea was higher in the loperamide-treated cohort.
Warnings and precautions
Neratinib is marketed as Nerlynx by Puma Biotechnology Inc. The prescribing information describes warnings and precautions relating to diarrhea, hepatotoxicity, and embryofetal toxicity. Patients should be monitored for diarrhea and treated with antidiarrheals as needed. Severe diarrhea with dehydration should be treated with fluids and electrolytes as needed, treatment should be interrupted and resumed at a reduced dose. For grade 3/4 diarrhea or diarrhea with complicating features (eg, dehydration, fever, neutropenia), stool cultures should be performed to rule out infectious causes.
Total bilirubin, aspartate and alanine aminotransferase, and alkaline phosphatase levels should be measured before starting treatment, every 3 months during treatment, or as clinically indicated. Neratinib can cause fetal harm, so pregnant women should be advised of the risk to the fetus and patients of reproductive potential should be counseled on the need for effective contraception during treatment and for at least 1 month after the last dose.4
The small-molecule tyrosine kinase inhibitor neratinib is now approved for the extended adjuvant treatment of patients with early-stage HER2 [human epidermal growth factor receptor]-positive breast cancer following postoperative trastuzumab. Trastuzumab is a HER2-targeted monoclonal antibody that has become standard of care in combination with chemotherapy for the treatment of this patient population in which it significantly improves survival. However, disease recurrence will occur in about a quarter of trastuzumab-treated patients owing to the development of resistance.
Neratinib may help overcome trastuzumab resistance thanks to its potent inhibition of the downstream phosphorylation of HER2 and other members of the HER family. Its approval was based on the phase 3 ExteNET trial, in which extended adjuvant treatment with neratinib was compared with placebo among 2,840 patients who remained disease free after 1 year of adjuvant trastuzumab.1
The ExteNET trial was performed at 495 centers in Europe, Asia, Australia, New Zealand, and South America. Patients aged 18 years or older (≥20 years in Japan), with stage 1-3 HER2-positive breast cancer, who completed neoadjuvant and adjuvant trastuzumab therapy up to 1 year before randomization were eligible. Patients also had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 (range, 0-5; 0, fully active, and 5, dead), normal organ function, and a left ventricular ejection fraction within normal institutional range. Patients with clinically significant cardiac, gastrointesintal or psychiatric comorbidities and those who were not able to swallow oral medication were excluded from the study.
Patients randomly received oral neratinib 240 mg per day or matching placebo, and randomization was stratified according to HR status (positive or negative), nodal status (0, 1-3, or ≥4) and trastuzumab-adjuvant regimen (sequentially or concurrently with chemotherapy).
The primary outcome was invasive disease-free survival (iDFS). The 2-year iDFS rate was 93.9% for neratinib, compared with 91.6% for placebo (hazard ratio [HR], 0.66; P < .008). Recently, a 5-year analysis of the ExteNET trial showed that after a median follow-up of 5.2 years, the iDFS rates were 90.2% vs 87.7% (HR, 0.73; P = .0083).2
Adverse events
The most common adverse event (AE) was diarrhea, in 95% of patients, 40% of whom had grade 3 diarrhea, leading to dose reduction in 26% of patients and discontinuation in 16.8% of patients. Serious AEs occurred in 7% of patients in the neratinib and 6% of those in the placebo arms. In the 5-year analysis, there was no evidence of increased risk of long-term toxicity or adverse consequences of neratinib-associated diarrhea. Furthermore, the ongoing, open-label phase 2 CONTROL trial suggests that the occurrence and severity of neratinib-associated diarrhea can be effectively controlled with antidiarrheal prophylaxis, with drugs such as loperamide.3
At the January 2017 cut-off, 137 patients treated with neratinib (240 mg/day) for 1 year had also received treatment with loperamide monotherapy, 64 patients had received loperamide and budesonide, and 10 patients had received loperamide and colestipol. The safety data from the loperamide monotherapy arm were compared with the safety data from the ExteNET trial, which was based in a similar population of patients who did not receive antidiarrheal prophylaxis. The incidence of all-grade diarrhea was 77% vs 95%, respectively, for those who received antidiarrheal prophylaxis in the CONTROL trial compared with those in the ExteNET trial who did not, and the repective rates of grade 3 diarrhea were 31% and 40%. The rate of dose reductions and holds owing to diarrhea were also lower among those who received antidiarrheal prophylaxis, but the rate of discontinuation due to diarrhea was higher in the loperamide-treated cohort.
Warnings and precautions
Neratinib is marketed as Nerlynx by Puma Biotechnology Inc. The prescribing information describes warnings and precautions relating to diarrhea, hepatotoxicity, and embryofetal toxicity. Patients should be monitored for diarrhea and treated with antidiarrheals as needed. Severe diarrhea with dehydration should be treated with fluids and electrolytes as needed, treatment should be interrupted and resumed at a reduced dose. For grade 3/4 diarrhea or diarrhea with complicating features (eg, dehydration, fever, neutropenia), stool cultures should be performed to rule out infectious causes.
Total bilirubin, aspartate and alanine aminotransferase, and alkaline phosphatase levels should be measured before starting treatment, every 3 months during treatment, or as clinically indicated. Neratinib can cause fetal harm, so pregnant women should be advised of the risk to the fetus and patients of reproductive potential should be counseled on the need for effective contraception during treatment and for at least 1 month after the last dose.4
1. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17: 367-377.
2. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab- based adjuvant therapy in HER2-positive breast cancer (ExteNET): a 5-year analysis of a randomised, double-blind, placebo- controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1688-1700.
3. Ibrahim E, Tripathy D, Wilkinson M, et al. E£ects of adding budesonide or colestipol to loperamide prophylaxis on neratinib-associated diarrhea in patients (pts) with HER2+ early-stage breast cancer (EBC): The CONTROL trial. Cancer Res. 2017; 77(13 supplement): Abstract CT128.
4. Nerlynx (neratinib) tablets, for oral use. Prescribing information. Puma Biotechnology Inc. https://nerlynx.com/pdf/full-prescribinginformation. pdf. Revised July 2017. Accessed November 20th, 2017.
1. Chan A, Delaloge S, Holmes FA, et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17: 367-377.
2. Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab- based adjuvant therapy in HER2-positive breast cancer (ExteNET): a 5-year analysis of a randomised, double-blind, placebo- controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1688-1700.
3. Ibrahim E, Tripathy D, Wilkinson M, et al. E£ects of adding budesonide or colestipol to loperamide prophylaxis on neratinib-associated diarrhea in patients (pts) with HER2+ early-stage breast cancer (EBC): The CONTROL trial. Cancer Res. 2017; 77(13 supplement): Abstract CT128.
4. Nerlynx (neratinib) tablets, for oral use. Prescribing information. Puma Biotechnology Inc. https://nerlynx.com/pdf/full-prescribinginformation. pdf. Revised July 2017. Accessed November 20th, 2017.
More biosimilars reach the market in efforts to improve access and cut costs
Biosimilars are copies of FDA-approved biologic drugs (those generally derived from a living organism) that cannot be identical to the reference drug but demonstrate a high similarity to it. As patents on the reference drugs expire, biosimilars are being developed to increase competition in the marketplace to reduce costs and improve patient access to therapy. Although the US Food and Drug Administration (FDA) has no regulatory power over drug prices, it recently announced efforts to streamline the biosimilar approval process to facilitate access to therapies and curb the associated skyrocketing costs.
Several biosimilars have been approved by the agency in recent years, and earlier this year they were joined by 2 more: the approval in May of epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for all indications of the reference product (epoetin alfa; Epogen/Procrit, Amgen), including the treatment of anemia caused by myelosuppressive chemotherapy, when there is a minimum of 2 additional months of planned chemotherapy;1 and the June approval of pegfilgrastim-jmdb (Fulphila, Mylan and Biocon) for the treatment of patients undergoing myelosuppressive chemotherapy to help reduce the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells).2 The reference product for pegfilgrastim-jmdb is pegfilgrastim (Neulasta, Amgen).
The approval of both biosimilars was based on a review of a body of evidence that included structural and functional characterization, animal study data, human pharmacokinetic (PK) and pharmacodynamic (PD) data, clinical immunogenicity data, and other clinical safety and efficacy data. This evidence established that the biosimilars were highly similar to the already FDA-approved reference products, with no clinically relevant differences.
Biocon and Mylan-GmBH, which jointly developed pegfilgrastim-jmdb, originally filed for approval in 2017; and Hospira Inc, a Pfizer company that developed epoetin alfa-epbx, filed for the first time in 2015. They subsequently received complete response letters from the FDA, twice in the case of the epoetin alfa biosimilar, rejecting their approval. For pegfilgrastim-jmdb, the complete response letter was related to a pending update of the Biologic License Application as the result of requalification activities taken because of modifications at their manufacturing plant. For epoetin alfa-epbx, the FDA expressed concerns relating to a manufacturing facility. The companies addressed the concerns in the complete response letters and submitted corrective and preventive action plans.3,4
Pegfilgrastim-jmdb
The results from a phase 3, multicenter, randomized, double-blind parallel-group trial of pegfilgrastim-jmdb compared with European Union-approved pegfilgrastim were published in 2016. Chemotherapy and radiation-naïve patients with newly diagnosed breast cancer (n = 194) received the biosimilar or reference product every 3 weeks for 6 cycles. The primary endpoint was duration of severe neutropenia in cycle 1, defined as days with absolute neutrophil count <0.5 x 109/L. The mean standard deviation was 1.2 [0.93] in the pegfilgrastim-jmdb arm and 1.2 [1.10] in the EU-pegfilgrastim arm, and the 95% confidence interval of least squares means differences was within the -1 day, +1 day range, indicating equivalency.5
A characterization and similarity assessment of pegfilgrastim-jmdb compared with US- and EU-approved pegfilgrastim was presented at the 2018 Annual Meeting of the American Society of Clinical Oncology. G-CSF receptor (G-CSFR) binding was assessed by surface plasmon resonance and potency was measured by in vitro stimulated proliferation in a mouse myelogenous leukemia cell line. In vivo rodent studies were also performed and included a PD study with a single dose of up to 3 mg/kg.6
There was high similarity in the structure, molecular mass, impurities and functional activity of the biosimilar and reference products, as well as similar G-CSFR binding and equivalent relative potency. Neutrophil and leukocyte counts were increased to a similar degree, and toxicology and drug kinetics were also comparable.
The recommended dose of pegfilgrastim-jmdb is a 6 mg/0.6 ml injection in a single-dose prefilled syringe for manual use only, administered subcutaneously once per chemotherapy cycle. The prescribing information also has dosing guidelines for administration in pediatric patients who weigh less than 45 kg. Pegfilgrastim-jmdb should not be administered between 14 days before and 24 hours after administration of chemotherapy.
The prescribing information details warnings and precautions relating to splenic rupture, acute respiratory distress syndrome (ARDS), serious allergic reactions, potential for severe/fatal sickle cell crises in patients with sickle cell disorders, glomerulonephritis, leukocytosis, capillary leak syndrome, and the potential for tumor growth or recurrence.7
Patients should be evaluated for an enlarged spleen or splenic rupture if they report upper left abdominal or shoulder pain. Patients who develop fever and lung infiltrates or respiratory distress should be evaluated for ARDS and treatment discontinued if a diagnosis is confirmed. Pegfilgrastim-jmdb should be permanently discontinued in patients who develop serious allergic reactions and should not be used in patients with a history of serious allergic reactions to pegfilgrastim or filgrastim products.
Dose-reduction or interruption should be considered in patients who develop glomerulonephritis. Complete blood counts should be monitored throughout treatment. Patients should be monitored closely for capillary leak syndrome and treated with standard therapy. Pegfilgrastim-jmdb is marketed as Fulphila.
Epoetin alfa-epbx
Epoetin alfa-epbx was evaluated in 2 clinical trials in healthy individuals. The EPOE-12-02 trial established the PK and PD following a single subcutaneous dose of 100 U/kg in 81 participants. The EPOE-14-1 study was designed to determine the PK and PD of multiple doses of subcutaneous 100 U/kg 3 times weekly for 3 weeks in 129 participants. Both studies met prespecified criteria
Evidence of efficacy and safety were also evaluated using pooled data from EPOE-10-13 and EPOE-10-01, conducted in patients with chronic kidney disease, which was considered the most sensitive population in which to evaluate clinically meaningful differences between the biosimilar and reference product.8,9
There were no clinically meaningful differences in efficacy and a similar adverse event profile. The most common side effects include high blood pressure, joint pain, muscle spasm, fever, dizziness, respiratory infection, and cough, among others.
The recommended dose of epoetin alfa-epbx, which is marketed as Retacrit, is 40,000 Units weekly or 150 U/kg 3 times weekly in adults and 600 U/kg intravenously weekly in pediatric patients aged 5 years or younger. Epoetin alfa-epbx comes with a boxed warning to alert health care providers to the increased risks of death, heart problems, stroke, and tumor growth, or recurrence. The prescribing information also details warnings and precautions relating to these risks, as well as hypertension, seizures, lack or loss of hemoglobin response, pure red cell aplasia, serious allergic reactions, and severe cutaneous reactions.9
Blood pressure should be appropriately controlled before treatment initiation, treatment should be reduced or withheld if it becomes uncontrollable, and patients should be advised of the importance of compliance with anti-hypertensive medication and dietary restrictions. Patients should be monitored closely for premonitory neurologic symptoms and advised to contact their provider in the event of new-onset seizures, premonitory symptoms, or change in seizure frequency.
The prescribing information has dosing recommendations for lack or loss of hemoglobin response to epoetin alfa-epbx. If severe anemia or low reticulocyte count occur, treatment should be withheld and patients evaluated for neutralizing antibodies to erythropoietin and, in the event that PRCA is confirmed, treatment should be permanently discontinued. Treatment should be immediately and permanently discontinued for serious allergic reactions or severe cutaneous reactions.
1. US Food and Drug Administration website. FDA approves first epoetin alfa biosimilar for the treatment of anemia. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm607703.htm. Updated May 15, 2018. Accessed June 22, 2018.
2. US Food and Drug Administration website. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm609805.htm. Updated June 4, 2018. Accessed June 22, 2018.
3. Reuters. BRIEF – Biocon says US FDA issues complete response letter for proposed biosimilar pegfilgrastim. https://www.reuters.com/article/brief-biocon-says-us-fda-issued-complete/brief-biocon-says-us-fda-issued-complete-response-letter-for-proposed-biosimilar-pegfilgrastim-idUSFWN1MK0Q1. Updated October 9, 2017. Accessed June 22, 2018.
4. FiercePharma. Pfizer, on third try, wins nod for biosimilar of blockbuster epogen/procrit. https://www.fiercepharma.com/pharma/pfizer-third-try-wins-fda-nod-for-biosimilar-blockbuster-epogen-procrit. Updated May 15, 2018. Accessed June 22, 2018.
5. Waller CF, Blakeley C, Pennella E. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):1433O.
6. Sankaran PV, Palanivelu DV, Nair R, et al. Characterization and similarity assessment of a pegfilgrastim biosimilar MYL-1401H. J Clin Oncol. 2018;36(suppl; abstr e19028).
7. Fulphila (pegfilgrastim-jmdb) injection, for subcutaneous use. Prescribing information. Mylan GmBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761075s000lbl.pdf. Released June 2018. Accessed June 22, 2018.
8. US Food and Drug Administration website. ‘Epoetin Hospira,’ a proposed biosimilar to US-licensed Epogen/Procrit. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Updated May 25, 2017. Accessed June 22, 2018.
9. Retacrit (epoetin alfa-epbx) injection, for intravenous or subcutaneous use. Prescribing information. Pfizer. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125545s000lbl.pdf. Released May 2018. Accessed June 22, 2018.
Biosimilars are copies of FDA-approved biologic drugs (those generally derived from a living organism) that cannot be identical to the reference drug but demonstrate a high similarity to it. As patents on the reference drugs expire, biosimilars are being developed to increase competition in the marketplace to reduce costs and improve patient access to therapy. Although the US Food and Drug Administration (FDA) has no regulatory power over drug prices, it recently announced efforts to streamline the biosimilar approval process to facilitate access to therapies and curb the associated skyrocketing costs.
Several biosimilars have been approved by the agency in recent years, and earlier this year they were joined by 2 more: the approval in May of epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for all indications of the reference product (epoetin alfa; Epogen/Procrit, Amgen), including the treatment of anemia caused by myelosuppressive chemotherapy, when there is a minimum of 2 additional months of planned chemotherapy;1 and the June approval of pegfilgrastim-jmdb (Fulphila, Mylan and Biocon) for the treatment of patients undergoing myelosuppressive chemotherapy to help reduce the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells).2 The reference product for pegfilgrastim-jmdb is pegfilgrastim (Neulasta, Amgen).
The approval of both biosimilars was based on a review of a body of evidence that included structural and functional characterization, animal study data, human pharmacokinetic (PK) and pharmacodynamic (PD) data, clinical immunogenicity data, and other clinical safety and efficacy data. This evidence established that the biosimilars were highly similar to the already FDA-approved reference products, with no clinically relevant differences.
Biocon and Mylan-GmBH, which jointly developed pegfilgrastim-jmdb, originally filed for approval in 2017; and Hospira Inc, a Pfizer company that developed epoetin alfa-epbx, filed for the first time in 2015. They subsequently received complete response letters from the FDA, twice in the case of the epoetin alfa biosimilar, rejecting their approval. For pegfilgrastim-jmdb, the complete response letter was related to a pending update of the Biologic License Application as the result of requalification activities taken because of modifications at their manufacturing plant. For epoetin alfa-epbx, the FDA expressed concerns relating to a manufacturing facility. The companies addressed the concerns in the complete response letters and submitted corrective and preventive action plans.3,4
Pegfilgrastim-jmdb
The results from a phase 3, multicenter, randomized, double-blind parallel-group trial of pegfilgrastim-jmdb compared with European Union-approved pegfilgrastim were published in 2016. Chemotherapy and radiation-naïve patients with newly diagnosed breast cancer (n = 194) received the biosimilar or reference product every 3 weeks for 6 cycles. The primary endpoint was duration of severe neutropenia in cycle 1, defined as days with absolute neutrophil count <0.5 x 109/L. The mean standard deviation was 1.2 [0.93] in the pegfilgrastim-jmdb arm and 1.2 [1.10] in the EU-pegfilgrastim arm, and the 95% confidence interval of least squares means differences was within the -1 day, +1 day range, indicating equivalency.5
A characterization and similarity assessment of pegfilgrastim-jmdb compared with US- and EU-approved pegfilgrastim was presented at the 2018 Annual Meeting of the American Society of Clinical Oncology. G-CSF receptor (G-CSFR) binding was assessed by surface plasmon resonance and potency was measured by in vitro stimulated proliferation in a mouse myelogenous leukemia cell line. In vivo rodent studies were also performed and included a PD study with a single dose of up to 3 mg/kg.6
There was high similarity in the structure, molecular mass, impurities and functional activity of the biosimilar and reference products, as well as similar G-CSFR binding and equivalent relative potency. Neutrophil and leukocyte counts were increased to a similar degree, and toxicology and drug kinetics were also comparable.
The recommended dose of pegfilgrastim-jmdb is a 6 mg/0.6 ml injection in a single-dose prefilled syringe for manual use only, administered subcutaneously once per chemotherapy cycle. The prescribing information also has dosing guidelines for administration in pediatric patients who weigh less than 45 kg. Pegfilgrastim-jmdb should not be administered between 14 days before and 24 hours after administration of chemotherapy.
The prescribing information details warnings and precautions relating to splenic rupture, acute respiratory distress syndrome (ARDS), serious allergic reactions, potential for severe/fatal sickle cell crises in patients with sickle cell disorders, glomerulonephritis, leukocytosis, capillary leak syndrome, and the potential for tumor growth or recurrence.7
Patients should be evaluated for an enlarged spleen or splenic rupture if they report upper left abdominal or shoulder pain. Patients who develop fever and lung infiltrates or respiratory distress should be evaluated for ARDS and treatment discontinued if a diagnosis is confirmed. Pegfilgrastim-jmdb should be permanently discontinued in patients who develop serious allergic reactions and should not be used in patients with a history of serious allergic reactions to pegfilgrastim or filgrastim products.
Dose-reduction or interruption should be considered in patients who develop glomerulonephritis. Complete blood counts should be monitored throughout treatment. Patients should be monitored closely for capillary leak syndrome and treated with standard therapy. Pegfilgrastim-jmdb is marketed as Fulphila.
Epoetin alfa-epbx
Epoetin alfa-epbx was evaluated in 2 clinical trials in healthy individuals. The EPOE-12-02 trial established the PK and PD following a single subcutaneous dose of 100 U/kg in 81 participants. The EPOE-14-1 study was designed to determine the PK and PD of multiple doses of subcutaneous 100 U/kg 3 times weekly for 3 weeks in 129 participants. Both studies met prespecified criteria
Evidence of efficacy and safety were also evaluated using pooled data from EPOE-10-13 and EPOE-10-01, conducted in patients with chronic kidney disease, which was considered the most sensitive population in which to evaluate clinically meaningful differences between the biosimilar and reference product.8,9
There were no clinically meaningful differences in efficacy and a similar adverse event profile. The most common side effects include high blood pressure, joint pain, muscle spasm, fever, dizziness, respiratory infection, and cough, among others.
The recommended dose of epoetin alfa-epbx, which is marketed as Retacrit, is 40,000 Units weekly or 150 U/kg 3 times weekly in adults and 600 U/kg intravenously weekly in pediatric patients aged 5 years or younger. Epoetin alfa-epbx comes with a boxed warning to alert health care providers to the increased risks of death, heart problems, stroke, and tumor growth, or recurrence. The prescribing information also details warnings and precautions relating to these risks, as well as hypertension, seizures, lack or loss of hemoglobin response, pure red cell aplasia, serious allergic reactions, and severe cutaneous reactions.9
Blood pressure should be appropriately controlled before treatment initiation, treatment should be reduced or withheld if it becomes uncontrollable, and patients should be advised of the importance of compliance with anti-hypertensive medication and dietary restrictions. Patients should be monitored closely for premonitory neurologic symptoms and advised to contact their provider in the event of new-onset seizures, premonitory symptoms, or change in seizure frequency.
The prescribing information has dosing recommendations for lack or loss of hemoglobin response to epoetin alfa-epbx. If severe anemia or low reticulocyte count occur, treatment should be withheld and patients evaluated for neutralizing antibodies to erythropoietin and, in the event that PRCA is confirmed, treatment should be permanently discontinued. Treatment should be immediately and permanently discontinued for serious allergic reactions or severe cutaneous reactions.
Biosimilars are copies of FDA-approved biologic drugs (those generally derived from a living organism) that cannot be identical to the reference drug but demonstrate a high similarity to it. As patents on the reference drugs expire, biosimilars are being developed to increase competition in the marketplace to reduce costs and improve patient access to therapy. Although the US Food and Drug Administration (FDA) has no regulatory power over drug prices, it recently announced efforts to streamline the biosimilar approval process to facilitate access to therapies and curb the associated skyrocketing costs.
Several biosimilars have been approved by the agency in recent years, and earlier this year they were joined by 2 more: the approval in May of epoetin alfa-epbx (Retacrit; Hospira, a Pfizer company) for all indications of the reference product (epoetin alfa; Epogen/Procrit, Amgen), including the treatment of anemia caused by myelosuppressive chemotherapy, when there is a minimum of 2 additional months of planned chemotherapy;1 and the June approval of pegfilgrastim-jmdb (Fulphila, Mylan and Biocon) for the treatment of patients undergoing myelosuppressive chemotherapy to help reduce the chance of infection as suggested by febrile neutropenia (fever, often with other signs of infection, associated with an abnormally low number of infection-fighting white blood cells).2 The reference product for pegfilgrastim-jmdb is pegfilgrastim (Neulasta, Amgen).
The approval of both biosimilars was based on a review of a body of evidence that included structural and functional characterization, animal study data, human pharmacokinetic (PK) and pharmacodynamic (PD) data, clinical immunogenicity data, and other clinical safety and efficacy data. This evidence established that the biosimilars were highly similar to the already FDA-approved reference products, with no clinically relevant differences.
Biocon and Mylan-GmBH, which jointly developed pegfilgrastim-jmdb, originally filed for approval in 2017; and Hospira Inc, a Pfizer company that developed epoetin alfa-epbx, filed for the first time in 2015. They subsequently received complete response letters from the FDA, twice in the case of the epoetin alfa biosimilar, rejecting their approval. For pegfilgrastim-jmdb, the complete response letter was related to a pending update of the Biologic License Application as the result of requalification activities taken because of modifications at their manufacturing plant. For epoetin alfa-epbx, the FDA expressed concerns relating to a manufacturing facility. The companies addressed the concerns in the complete response letters and submitted corrective and preventive action plans.3,4
Pegfilgrastim-jmdb
The results from a phase 3, multicenter, randomized, double-blind parallel-group trial of pegfilgrastim-jmdb compared with European Union-approved pegfilgrastim were published in 2016. Chemotherapy and radiation-naïve patients with newly diagnosed breast cancer (n = 194) received the biosimilar or reference product every 3 weeks for 6 cycles. The primary endpoint was duration of severe neutropenia in cycle 1, defined as days with absolute neutrophil count <0.5 x 109/L. The mean standard deviation was 1.2 [0.93] in the pegfilgrastim-jmdb arm and 1.2 [1.10] in the EU-pegfilgrastim arm, and the 95% confidence interval of least squares means differences was within the -1 day, +1 day range, indicating equivalency.5
A characterization and similarity assessment of pegfilgrastim-jmdb compared with US- and EU-approved pegfilgrastim was presented at the 2018 Annual Meeting of the American Society of Clinical Oncology. G-CSF receptor (G-CSFR) binding was assessed by surface plasmon resonance and potency was measured by in vitro stimulated proliferation in a mouse myelogenous leukemia cell line. In vivo rodent studies were also performed and included a PD study with a single dose of up to 3 mg/kg.6
There was high similarity in the structure, molecular mass, impurities and functional activity of the biosimilar and reference products, as well as similar G-CSFR binding and equivalent relative potency. Neutrophil and leukocyte counts were increased to a similar degree, and toxicology and drug kinetics were also comparable.
The recommended dose of pegfilgrastim-jmdb is a 6 mg/0.6 ml injection in a single-dose prefilled syringe for manual use only, administered subcutaneously once per chemotherapy cycle. The prescribing information also has dosing guidelines for administration in pediatric patients who weigh less than 45 kg. Pegfilgrastim-jmdb should not be administered between 14 days before and 24 hours after administration of chemotherapy.
The prescribing information details warnings and precautions relating to splenic rupture, acute respiratory distress syndrome (ARDS), serious allergic reactions, potential for severe/fatal sickle cell crises in patients with sickle cell disorders, glomerulonephritis, leukocytosis, capillary leak syndrome, and the potential for tumor growth or recurrence.7
Patients should be evaluated for an enlarged spleen or splenic rupture if they report upper left abdominal or shoulder pain. Patients who develop fever and lung infiltrates or respiratory distress should be evaluated for ARDS and treatment discontinued if a diagnosis is confirmed. Pegfilgrastim-jmdb should be permanently discontinued in patients who develop serious allergic reactions and should not be used in patients with a history of serious allergic reactions to pegfilgrastim or filgrastim products.
Dose-reduction or interruption should be considered in patients who develop glomerulonephritis. Complete blood counts should be monitored throughout treatment. Patients should be monitored closely for capillary leak syndrome and treated with standard therapy. Pegfilgrastim-jmdb is marketed as Fulphila.
Epoetin alfa-epbx
Epoetin alfa-epbx was evaluated in 2 clinical trials in healthy individuals. The EPOE-12-02 trial established the PK and PD following a single subcutaneous dose of 100 U/kg in 81 participants. The EPOE-14-1 study was designed to determine the PK and PD of multiple doses of subcutaneous 100 U/kg 3 times weekly for 3 weeks in 129 participants. Both studies met prespecified criteria
Evidence of efficacy and safety were also evaluated using pooled data from EPOE-10-13 and EPOE-10-01, conducted in patients with chronic kidney disease, which was considered the most sensitive population in which to evaluate clinically meaningful differences between the biosimilar and reference product.8,9
There were no clinically meaningful differences in efficacy and a similar adverse event profile. The most common side effects include high blood pressure, joint pain, muscle spasm, fever, dizziness, respiratory infection, and cough, among others.
The recommended dose of epoetin alfa-epbx, which is marketed as Retacrit, is 40,000 Units weekly or 150 U/kg 3 times weekly in adults and 600 U/kg intravenously weekly in pediatric patients aged 5 years or younger. Epoetin alfa-epbx comes with a boxed warning to alert health care providers to the increased risks of death, heart problems, stroke, and tumor growth, or recurrence. The prescribing information also details warnings and precautions relating to these risks, as well as hypertension, seizures, lack or loss of hemoglobin response, pure red cell aplasia, serious allergic reactions, and severe cutaneous reactions.9
Blood pressure should be appropriately controlled before treatment initiation, treatment should be reduced or withheld if it becomes uncontrollable, and patients should be advised of the importance of compliance with anti-hypertensive medication and dietary restrictions. Patients should be monitored closely for premonitory neurologic symptoms and advised to contact their provider in the event of new-onset seizures, premonitory symptoms, or change in seizure frequency.
The prescribing information has dosing recommendations for lack or loss of hemoglobin response to epoetin alfa-epbx. If severe anemia or low reticulocyte count occur, treatment should be withheld and patients evaluated for neutralizing antibodies to erythropoietin and, in the event that PRCA is confirmed, treatment should be permanently discontinued. Treatment should be immediately and permanently discontinued for serious allergic reactions or severe cutaneous reactions.
1. US Food and Drug Administration website. FDA approves first epoetin alfa biosimilar for the treatment of anemia. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm607703.htm. Updated May 15, 2018. Accessed June 22, 2018.
2. US Food and Drug Administration website. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm609805.htm. Updated June 4, 2018. Accessed June 22, 2018.
3. Reuters. BRIEF – Biocon says US FDA issues complete response letter for proposed biosimilar pegfilgrastim. https://www.reuters.com/article/brief-biocon-says-us-fda-issued-complete/brief-biocon-says-us-fda-issued-complete-response-letter-for-proposed-biosimilar-pegfilgrastim-idUSFWN1MK0Q1. Updated October 9, 2017. Accessed June 22, 2018.
4. FiercePharma. Pfizer, on third try, wins nod for biosimilar of blockbuster epogen/procrit. https://www.fiercepharma.com/pharma/pfizer-third-try-wins-fda-nod-for-biosimilar-blockbuster-epogen-procrit. Updated May 15, 2018. Accessed June 22, 2018.
5. Waller CF, Blakeley C, Pennella E. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):1433O.
6. Sankaran PV, Palanivelu DV, Nair R, et al. Characterization and similarity assessment of a pegfilgrastim biosimilar MYL-1401H. J Clin Oncol. 2018;36(suppl; abstr e19028).
7. Fulphila (pegfilgrastim-jmdb) injection, for subcutaneous use. Prescribing information. Mylan GmBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761075s000lbl.pdf. Released June 2018. Accessed June 22, 2018.
8. US Food and Drug Administration website. ‘Epoetin Hospira,’ a proposed biosimilar to US-licensed Epogen/Procrit. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Updated May 25, 2017. Accessed June 22, 2018.
9. Retacrit (epoetin alfa-epbx) injection, for intravenous or subcutaneous use. Prescribing information. Pfizer. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125545s000lbl.pdf. Released May 2018. Accessed June 22, 2018.
1. US Food and Drug Administration website. FDA approves first epoetin alfa biosimilar for the treatment of anemia. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm607703.htm. Updated May 15, 2018. Accessed June 22, 2018.
2. US Food and Drug Administration website. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm609805.htm. Updated June 4, 2018. Accessed June 22, 2018.
3. Reuters. BRIEF – Biocon says US FDA issues complete response letter for proposed biosimilar pegfilgrastim. https://www.reuters.com/article/brief-biocon-says-us-fda-issued-complete/brief-biocon-says-us-fda-issued-complete-response-letter-for-proposed-biosimilar-pegfilgrastim-idUSFWN1MK0Q1. Updated October 9, 2017. Accessed June 22, 2018.
4. FiercePharma. Pfizer, on third try, wins nod for biosimilar of blockbuster epogen/procrit. https://www.fiercepharma.com/pharma/pfizer-third-try-wins-fda-nod-for-biosimilar-blockbuster-epogen-procrit. Updated May 15, 2018. Accessed June 22, 2018.
5. Waller CF, Blakeley C, Pennella E. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):1433O.
6. Sankaran PV, Palanivelu DV, Nair R, et al. Characterization and similarity assessment of a pegfilgrastim biosimilar MYL-1401H. J Clin Oncol. 2018;36(suppl; abstr e19028).
7. Fulphila (pegfilgrastim-jmdb) injection, for subcutaneous use. Prescribing information. Mylan GmBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761075s000lbl.pdf. Released June 2018. Accessed June 22, 2018.
8. US Food and Drug Administration website. ‘Epoetin Hospira,’ a proposed biosimilar to US-licensed Epogen/Procrit. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Updated May 25, 2017. Accessed June 22, 2018.
9. Retacrit (epoetin alfa-epbx) injection, for intravenous or subcutaneous use. Prescribing information. Pfizer. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125545s000lbl.pdf. Released May 2018. Accessed June 22, 2018.
First CAR T-cell therapy approvals bolster booming immunotherapy market
There were a number of landmark approvals by the US Food and Drug Administration (FDA) in 2017 for cancer therapies, among them, the approval of the first two chimeric antigen receptor (CAR) T-cell therapies for cancer: tisagenlecleucel (in August) and axicabtagene ciloluecel (in October).1 CAR T-cells are a type of adoptive cell therapy or immunotherapy, in which the patient’s own immune cells are genetically engineered to target a tumor-associated antigen, in this case CD19. In tisagenlecleucel, CD19 proteins on B cells are targeted in the treatment of B-cell precursor acute lymphoblastic leukemia. Axicabtagene ciloluecel, the second anti-CD19 CAR T-cell therapy, was approved for the treatment of refractory, aggressive B-cell non-Hodgkin lymphoma.
Tisagenlecleucel
Tisagenlecleucel was approved for the treatment of pediatric patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) whose disease is refractory to treatment or who have relapsed after second-line therapy or beyond.2 Approval was based on the pivotal ELIANA trial, a single-arm, global phase 2 trial conducted at 25 centers worldwide during April 2015 through April 2017. Patients were eligible for enrollment if they had relapsed or refractory B-cell ALL and were at least 3 years of age at screening and no older than 21 years of age at diagnosis, had at least 5% lymphoblasts in the bone marrow at screening, had tumor expression of CD19, had adequate organ function, and a Karnofsky (adult) or Lansky (child) Performance Status of ≥50 (with the worst allowable score, 50, indicating a patient who requires considerable assistance and frequent medical care [Karnofsky] and lying around much of the day, but gets dressed; no active playing but participates in all quiet play and activities [Lansky]). Exclusion criteria included previous receipt of anti-CD19 therapy, concomitant genetic syndromes associated with bone marrow failure, previous malignancy, and/or active or latent hepatitis B or C virus (HBV/HCV) infection.
The overall remission rate (ORR) was evaluated in 75 patients who were given a single dose of tisagenlecleucel (a median weight-adjusted dose of 3.1 x 106 transduced viable T cells per kg of body weight) within 14 days of completing a lymphodepleting chemotherapy regimen. The confirmed ORR after at least 3 months of follow-up, as assessed by independent central review, was 81%, which included 60% of patients in complete remission (CR) and 21% in complete remission with incomplete hematologic recovery, all of whom were negative for minimal residual disease.
The most common adverse events (AEs) associated with tisagenlecleucel treatment were cytokine release syndrome (CRS), hypogammaglobulinemia, infection, pyrexia, decreased appetite, headache, encephalopathy, hypotension, bleeding episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, and delirium. AEs were of grade 3/4 severity in 84% of patients.3
To combat serious safety issues, including CRS and neurologic toxicities, the FDA approved tisagenlecleucel with a Risk Evaluation and Mitigation Strategy (REMS) that, in part, requires health care providers who administer the drug to be trained in their management. It also requires the facility where treatment is administered to have immediate, onsite access to the drug tocilizumab, which was approved in conjunction with tisagenlecleucel for the treatment of patients who experience CRS.
In addition to information about the REMS, the prescribing information details warnings and precautions relating to several other common toxicities. These include hypersensitivity reactions, serious infections, prolonged cytopenias, and hypogammaglobulinemia.
Patients should be monitored for signs and symptoms of infection and treated appropriately. Viral reactivation can occur after tisagenlecleucel treatment, so patients should be screened for HBV, HCV, and human immunodeficiency virus before collection of cells.
The administration of myeloid growth factors is not recommended during the first 3 weeks after infusion or until CRS has resolved. Immunoglobulin levels should be monitored after treatment and hypogammaglobulinemia managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement according to standard guidelines.
Patients treated with tisagenlecleucel should also be monitored for life for secondary malignancies, should not be treated with live vaccines from 2 weeks before the start of lymphodepleting chemotherapy until immune recovery after tisagenlecleucel infusion, and should be aware of the potential for neurological events to impact their ability to drive and use dangerous machinery.4
Tisagenlecleucel is marketed as Kymriah by Novartis Pharmaceuticals. The recommended dose is 1 infusion of 0.2-5 x 106 CAR-positive viable T cells per kilogram of body weight intravenously (for patients ≤50kg) and 0.1-2.5 x 108 cells/kg (for patients >50kg), administered 2-14 days after lymphodepleting chemotherapy.
Axicabtagene ciloleucel
Axicabtagene ciloleucel was approved for the treatment of adult patients with certain types of relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.5 It is not indicated for the treatment of patients with primary central nervous system lymphoma.
Approval followed positive results from the phase 2 single-arm, multicenter ZUMA-1 trial.6 Patients were included if they were aged 18 years of age and older, had histologically confirmed aggressive B-cell non-Hodgkin lymphoma that was chemotherapy refractory, had received adequate previous therapy, had at least 1 measurable lesion, had completed radiation or systemic therapy at least 2 weeks before, had resolved toxicities related to previous therapy, and had an Eastern Cooperative Oncology Group Performance Status of 0 (asymptomatic) or 1 (symptomatic), an absolute neutrophil count of ≥1000/µL, a platelet count of ≥50,000/µL, and adequate hepatic, renal and cardiac function. They were treated with a single infusion of axicabtagene ciloleucel after lymphodepleting chemotherapy.
Patients who had received previous CD19-targeted therapy, who had concomitant genetic syndromes associated with bone marrow failure, who had previous malignancy, and who had active or latent HBV/HCV infection were among those excluded from the study.
Patients were enrolled in 2 cohorts; those with DLBCL (n = 77) and those with PMBCL or transformed follicular lymphoma (n = 24). The primary endpoint was objective response rate, and after a primary analysis at a minimum of 6 months follow-up, the objective response rate was 82%, with a CR rate of 52%. Among patients who achieved CR, the median duration of response was not reached after a median follow-up of 7.9 months.
A subsequent updated analysis was performed when 108 patients had been followed for a minimum of 1 year. The objective response rate was 82%, and the CR rate was 58%, with some patients having CR in the absence of additional therapies as late as 15 months after treatment. At this updated analysis, 42% of patients continued to have a response, 40% of whom remained in CR.
The most common grade 3 or higher AEs included febrile neutropenia, fever, CRS, encephalopathy, infections, hypotension, and hypoxia. Serious AEs occurred in 52% of patients and included CRS, neurologic toxicity, prolonged cytopenias, and serious infections. Grade 3 or higher CRS or neurologic toxicities occurred in 13% and 28% of patients, respectively. Three patients died during treatment.
To mitigate the risk of CRS and neurologic toxicity, axicabtagene ciloleucel is approved with an REMS that requires appropriate certification and training before hospitals are cleared to administer the therapy.
Other warnings and precautions in the prescribing information relate to serious infections (monitor for signs and symptoms and treat appropriately), prolonged cytopenias (monitor blood counts), hypogammaglobulinemia (monitor immunoglobulin levels and manage appropriately), secondary malignancies (life-long monitoring), and the potential effects of neurologic events on a patient’s ability to drive and operate dangerous machinery (avoid for at least 8 weeks after infusion).7
Axicabtagene ciloleucel is marketed as Yescarta by Kite Pharma Inc. The recommended dose is a single intravenous infusion with a target of 2 x 106 CAR-positive viable T cells per kilogram of body weight, preceded by fludarabine and cyclophosphamide lymphodepleting chemotherapy.
1. Bosserman LD. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. JCSO 2017;15(6):e283-e290.
2. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA News Release. August 30, 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/
ucm574154.htm. Accessed March 31, 2018.
3. Maude S.L, Laetsch T.W, Buechner S, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48.
4. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Novartis Pharmaceuticals Corporation, August, 2017. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.
com/files/kymriah.pdf. Accessed March 31, 2018.
5. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA News Release. October 18, 2017. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Accessed March 31, 2018.
6. Neelapu, S.S, Locke F.L, Bartlett, L.J, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531-44.
7. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Kite Pharma Inc. October 2017. https://www.yescarta.com/wp-content/uploads/yescarta-pi.pdf. Accessed March 31, 2018.
There were a number of landmark approvals by the US Food and Drug Administration (FDA) in 2017 for cancer therapies, among them, the approval of the first two chimeric antigen receptor (CAR) T-cell therapies for cancer: tisagenlecleucel (in August) and axicabtagene ciloluecel (in October).1 CAR T-cells are a type of adoptive cell therapy or immunotherapy, in which the patient’s own immune cells are genetically engineered to target a tumor-associated antigen, in this case CD19. In tisagenlecleucel, CD19 proteins on B cells are targeted in the treatment of B-cell precursor acute lymphoblastic leukemia. Axicabtagene ciloluecel, the second anti-CD19 CAR T-cell therapy, was approved for the treatment of refractory, aggressive B-cell non-Hodgkin lymphoma.
Tisagenlecleucel
Tisagenlecleucel was approved for the treatment of pediatric patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) whose disease is refractory to treatment or who have relapsed after second-line therapy or beyond.2 Approval was based on the pivotal ELIANA trial, a single-arm, global phase 2 trial conducted at 25 centers worldwide during April 2015 through April 2017. Patients were eligible for enrollment if they had relapsed or refractory B-cell ALL and were at least 3 years of age at screening and no older than 21 years of age at diagnosis, had at least 5% lymphoblasts in the bone marrow at screening, had tumor expression of CD19, had adequate organ function, and a Karnofsky (adult) or Lansky (child) Performance Status of ≥50 (with the worst allowable score, 50, indicating a patient who requires considerable assistance and frequent medical care [Karnofsky] and lying around much of the day, but gets dressed; no active playing but participates in all quiet play and activities [Lansky]). Exclusion criteria included previous receipt of anti-CD19 therapy, concomitant genetic syndromes associated with bone marrow failure, previous malignancy, and/or active or latent hepatitis B or C virus (HBV/HCV) infection.
The overall remission rate (ORR) was evaluated in 75 patients who were given a single dose of tisagenlecleucel (a median weight-adjusted dose of 3.1 x 106 transduced viable T cells per kg of body weight) within 14 days of completing a lymphodepleting chemotherapy regimen. The confirmed ORR after at least 3 months of follow-up, as assessed by independent central review, was 81%, which included 60% of patients in complete remission (CR) and 21% in complete remission with incomplete hematologic recovery, all of whom were negative for minimal residual disease.
The most common adverse events (AEs) associated with tisagenlecleucel treatment were cytokine release syndrome (CRS), hypogammaglobulinemia, infection, pyrexia, decreased appetite, headache, encephalopathy, hypotension, bleeding episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, and delirium. AEs were of grade 3/4 severity in 84% of patients.3
To combat serious safety issues, including CRS and neurologic toxicities, the FDA approved tisagenlecleucel with a Risk Evaluation and Mitigation Strategy (REMS) that, in part, requires health care providers who administer the drug to be trained in their management. It also requires the facility where treatment is administered to have immediate, onsite access to the drug tocilizumab, which was approved in conjunction with tisagenlecleucel for the treatment of patients who experience CRS.
In addition to information about the REMS, the prescribing information details warnings and precautions relating to several other common toxicities. These include hypersensitivity reactions, serious infections, prolonged cytopenias, and hypogammaglobulinemia.
Patients should be monitored for signs and symptoms of infection and treated appropriately. Viral reactivation can occur after tisagenlecleucel treatment, so patients should be screened for HBV, HCV, and human immunodeficiency virus before collection of cells.
The administration of myeloid growth factors is not recommended during the first 3 weeks after infusion or until CRS has resolved. Immunoglobulin levels should be monitored after treatment and hypogammaglobulinemia managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement according to standard guidelines.
Patients treated with tisagenlecleucel should also be monitored for life for secondary malignancies, should not be treated with live vaccines from 2 weeks before the start of lymphodepleting chemotherapy until immune recovery after tisagenlecleucel infusion, and should be aware of the potential for neurological events to impact their ability to drive and use dangerous machinery.4
Tisagenlecleucel is marketed as Kymriah by Novartis Pharmaceuticals. The recommended dose is 1 infusion of 0.2-5 x 106 CAR-positive viable T cells per kilogram of body weight intravenously (for patients ≤50kg) and 0.1-2.5 x 108 cells/kg (for patients >50kg), administered 2-14 days after lymphodepleting chemotherapy.
Axicabtagene ciloleucel
Axicabtagene ciloleucel was approved for the treatment of adult patients with certain types of relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.5 It is not indicated for the treatment of patients with primary central nervous system lymphoma.
Approval followed positive results from the phase 2 single-arm, multicenter ZUMA-1 trial.6 Patients were included if they were aged 18 years of age and older, had histologically confirmed aggressive B-cell non-Hodgkin lymphoma that was chemotherapy refractory, had received adequate previous therapy, had at least 1 measurable lesion, had completed radiation or systemic therapy at least 2 weeks before, had resolved toxicities related to previous therapy, and had an Eastern Cooperative Oncology Group Performance Status of 0 (asymptomatic) or 1 (symptomatic), an absolute neutrophil count of ≥1000/µL, a platelet count of ≥50,000/µL, and adequate hepatic, renal and cardiac function. They were treated with a single infusion of axicabtagene ciloleucel after lymphodepleting chemotherapy.
Patients who had received previous CD19-targeted therapy, who had concomitant genetic syndromes associated with bone marrow failure, who had previous malignancy, and who had active or latent HBV/HCV infection were among those excluded from the study.
Patients were enrolled in 2 cohorts; those with DLBCL (n = 77) and those with PMBCL or transformed follicular lymphoma (n = 24). The primary endpoint was objective response rate, and after a primary analysis at a minimum of 6 months follow-up, the objective response rate was 82%, with a CR rate of 52%. Among patients who achieved CR, the median duration of response was not reached after a median follow-up of 7.9 months.
A subsequent updated analysis was performed when 108 patients had been followed for a minimum of 1 year. The objective response rate was 82%, and the CR rate was 58%, with some patients having CR in the absence of additional therapies as late as 15 months after treatment. At this updated analysis, 42% of patients continued to have a response, 40% of whom remained in CR.
The most common grade 3 or higher AEs included febrile neutropenia, fever, CRS, encephalopathy, infections, hypotension, and hypoxia. Serious AEs occurred in 52% of patients and included CRS, neurologic toxicity, prolonged cytopenias, and serious infections. Grade 3 or higher CRS or neurologic toxicities occurred in 13% and 28% of patients, respectively. Three patients died during treatment.
To mitigate the risk of CRS and neurologic toxicity, axicabtagene ciloleucel is approved with an REMS that requires appropriate certification and training before hospitals are cleared to administer the therapy.
Other warnings and precautions in the prescribing information relate to serious infections (monitor for signs and symptoms and treat appropriately), prolonged cytopenias (monitor blood counts), hypogammaglobulinemia (monitor immunoglobulin levels and manage appropriately), secondary malignancies (life-long monitoring), and the potential effects of neurologic events on a patient’s ability to drive and operate dangerous machinery (avoid for at least 8 weeks after infusion).7
Axicabtagene ciloleucel is marketed as Yescarta by Kite Pharma Inc. The recommended dose is a single intravenous infusion with a target of 2 x 106 CAR-positive viable T cells per kilogram of body weight, preceded by fludarabine and cyclophosphamide lymphodepleting chemotherapy.
There were a number of landmark approvals by the US Food and Drug Administration (FDA) in 2017 for cancer therapies, among them, the approval of the first two chimeric antigen receptor (CAR) T-cell therapies for cancer: tisagenlecleucel (in August) and axicabtagene ciloluecel (in October).1 CAR T-cells are a type of adoptive cell therapy or immunotherapy, in which the patient’s own immune cells are genetically engineered to target a tumor-associated antigen, in this case CD19. In tisagenlecleucel, CD19 proteins on B cells are targeted in the treatment of B-cell precursor acute lymphoblastic leukemia. Axicabtagene ciloluecel, the second anti-CD19 CAR T-cell therapy, was approved for the treatment of refractory, aggressive B-cell non-Hodgkin lymphoma.
Tisagenlecleucel
Tisagenlecleucel was approved for the treatment of pediatric patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) whose disease is refractory to treatment or who have relapsed after second-line therapy or beyond.2 Approval was based on the pivotal ELIANA trial, a single-arm, global phase 2 trial conducted at 25 centers worldwide during April 2015 through April 2017. Patients were eligible for enrollment if they had relapsed or refractory B-cell ALL and were at least 3 years of age at screening and no older than 21 years of age at diagnosis, had at least 5% lymphoblasts in the bone marrow at screening, had tumor expression of CD19, had adequate organ function, and a Karnofsky (adult) or Lansky (child) Performance Status of ≥50 (with the worst allowable score, 50, indicating a patient who requires considerable assistance and frequent medical care [Karnofsky] and lying around much of the day, but gets dressed; no active playing but participates in all quiet play and activities [Lansky]). Exclusion criteria included previous receipt of anti-CD19 therapy, concomitant genetic syndromes associated with bone marrow failure, previous malignancy, and/or active or latent hepatitis B or C virus (HBV/HCV) infection.
The overall remission rate (ORR) was evaluated in 75 patients who were given a single dose of tisagenlecleucel (a median weight-adjusted dose of 3.1 x 106 transduced viable T cells per kg of body weight) within 14 days of completing a lymphodepleting chemotherapy regimen. The confirmed ORR after at least 3 months of follow-up, as assessed by independent central review, was 81%, which included 60% of patients in complete remission (CR) and 21% in complete remission with incomplete hematologic recovery, all of whom were negative for minimal residual disease.
The most common adverse events (AEs) associated with tisagenlecleucel treatment were cytokine release syndrome (CRS), hypogammaglobulinemia, infection, pyrexia, decreased appetite, headache, encephalopathy, hypotension, bleeding episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, and delirium. AEs were of grade 3/4 severity in 84% of patients.3
To combat serious safety issues, including CRS and neurologic toxicities, the FDA approved tisagenlecleucel with a Risk Evaluation and Mitigation Strategy (REMS) that, in part, requires health care providers who administer the drug to be trained in their management. It also requires the facility where treatment is administered to have immediate, onsite access to the drug tocilizumab, which was approved in conjunction with tisagenlecleucel for the treatment of patients who experience CRS.
In addition to information about the REMS, the prescribing information details warnings and precautions relating to several other common toxicities. These include hypersensitivity reactions, serious infections, prolonged cytopenias, and hypogammaglobulinemia.
Patients should be monitored for signs and symptoms of infection and treated appropriately. Viral reactivation can occur after tisagenlecleucel treatment, so patients should be screened for HBV, HCV, and human immunodeficiency virus before collection of cells.
The administration of myeloid growth factors is not recommended during the first 3 weeks after infusion or until CRS has resolved. Immunoglobulin levels should be monitored after treatment and hypogammaglobulinemia managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement according to standard guidelines.
Patients treated with tisagenlecleucel should also be monitored for life for secondary malignancies, should not be treated with live vaccines from 2 weeks before the start of lymphodepleting chemotherapy until immune recovery after tisagenlecleucel infusion, and should be aware of the potential for neurological events to impact their ability to drive and use dangerous machinery.4
Tisagenlecleucel is marketed as Kymriah by Novartis Pharmaceuticals. The recommended dose is 1 infusion of 0.2-5 x 106 CAR-positive viable T cells per kilogram of body weight intravenously (for patients ≤50kg) and 0.1-2.5 x 108 cells/kg (for patients >50kg), administered 2-14 days after lymphodepleting chemotherapy.
Axicabtagene ciloleucel
Axicabtagene ciloleucel was approved for the treatment of adult patients with certain types of relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.5 It is not indicated for the treatment of patients with primary central nervous system lymphoma.
Approval followed positive results from the phase 2 single-arm, multicenter ZUMA-1 trial.6 Patients were included if they were aged 18 years of age and older, had histologically confirmed aggressive B-cell non-Hodgkin lymphoma that was chemotherapy refractory, had received adequate previous therapy, had at least 1 measurable lesion, had completed radiation or systemic therapy at least 2 weeks before, had resolved toxicities related to previous therapy, and had an Eastern Cooperative Oncology Group Performance Status of 0 (asymptomatic) or 1 (symptomatic), an absolute neutrophil count of ≥1000/µL, a platelet count of ≥50,000/µL, and adequate hepatic, renal and cardiac function. They were treated with a single infusion of axicabtagene ciloleucel after lymphodepleting chemotherapy.
Patients who had received previous CD19-targeted therapy, who had concomitant genetic syndromes associated with bone marrow failure, who had previous malignancy, and who had active or latent HBV/HCV infection were among those excluded from the study.
Patients were enrolled in 2 cohorts; those with DLBCL (n = 77) and those with PMBCL or transformed follicular lymphoma (n = 24). The primary endpoint was objective response rate, and after a primary analysis at a minimum of 6 months follow-up, the objective response rate was 82%, with a CR rate of 52%. Among patients who achieved CR, the median duration of response was not reached after a median follow-up of 7.9 months.
A subsequent updated analysis was performed when 108 patients had been followed for a minimum of 1 year. The objective response rate was 82%, and the CR rate was 58%, with some patients having CR in the absence of additional therapies as late as 15 months after treatment. At this updated analysis, 42% of patients continued to have a response, 40% of whom remained in CR.
The most common grade 3 or higher AEs included febrile neutropenia, fever, CRS, encephalopathy, infections, hypotension, and hypoxia. Serious AEs occurred in 52% of patients and included CRS, neurologic toxicity, prolonged cytopenias, and serious infections. Grade 3 or higher CRS or neurologic toxicities occurred in 13% and 28% of patients, respectively. Three patients died during treatment.
To mitigate the risk of CRS and neurologic toxicity, axicabtagene ciloleucel is approved with an REMS that requires appropriate certification and training before hospitals are cleared to administer the therapy.
Other warnings and precautions in the prescribing information relate to serious infections (monitor for signs and symptoms and treat appropriately), prolonged cytopenias (monitor blood counts), hypogammaglobulinemia (monitor immunoglobulin levels and manage appropriately), secondary malignancies (life-long monitoring), and the potential effects of neurologic events on a patient’s ability to drive and operate dangerous machinery (avoid for at least 8 weeks after infusion).7
Axicabtagene ciloleucel is marketed as Yescarta by Kite Pharma Inc. The recommended dose is a single intravenous infusion with a target of 2 x 106 CAR-positive viable T cells per kilogram of body weight, preceded by fludarabine and cyclophosphamide lymphodepleting chemotherapy.
1. Bosserman LD. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. JCSO 2017;15(6):e283-e290.
2. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA News Release. August 30, 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/
ucm574154.htm. Accessed March 31, 2018.
3. Maude S.L, Laetsch T.W, Buechner S, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48.
4. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Novartis Pharmaceuticals Corporation, August, 2017. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.
com/files/kymriah.pdf. Accessed March 31, 2018.
5. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA News Release. October 18, 2017. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Accessed March 31, 2018.
6. Neelapu, S.S, Locke F.L, Bartlett, L.J, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531-44.
7. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Kite Pharma Inc. October 2017. https://www.yescarta.com/wp-content/uploads/yescarta-pi.pdf. Accessed March 31, 2018.
1. Bosserman LD. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. JCSO 2017;15(6):e283-e290.
2. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA News Release. August 30, 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/
ucm574154.htm. Accessed March 31, 2018.
3. Maude S.L, Laetsch T.W, Buechner S, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48.
4. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Novartis Pharmaceuticals Corporation, August, 2017. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.
com/files/kymriah.pdf. Accessed March 31, 2018.
5. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA News Release. October 18, 2017. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Accessed March 31, 2018.
6. Neelapu, S.S, Locke F.L, Bartlett, L.J, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531-44.
7. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Kite Pharma Inc. October 2017. https://www.yescarta.com/wp-content/uploads/yescarta-pi.pdf. Accessed March 31, 2018.
Lenalidomide becomes standard of care for multiple myeloma in the maintenance setting
The treatment of multiple myeloma has been revolutionized in the past few decades, with the introduction of numerous novel drug classes that have more than doubled median survival times. The immunomodulatory drug (IMiD), lenalidomide, forms the backbone of the majority of treatment paradigms, first receiving US Food and Drug Administration approval in 2006 for use in combination with dexamethasone in previously treated patients with multiple myeloma. Since then, approved indications for lenalidomide in multiple myeloma have continued to expand.
Most recently, on February 22, 2017, lenalidomide was approved for use as maintenance therapy following autologous stem cell transplant (ASCT), making it the first and only treatment available in this setting. This approval was based on 2 randomized, controlled trials that evaluated the efficacy and safety of lenalidomide in more than 1,000 patients in this setting and demonstrated a significant advantage in progression-free survival (PFS) compared with patients receiving placebo.
CALGB 1001041 and IFM 2005-022 were randomized, double-blind phase 3 trials conducted at 47 locations across the United States and 78 centers in France, Belgium, and Switzerland, respectively. In the CALGB trial, eligible patients were 18-70 years of age, with a European Cooperative Oncology Group (ECOG) performance status of 0 or 1, symptomatic disease requiring treatment (Durie-Salmon stage ≥1), and who received any induction therapy of 2-12 months duration. In the IFM trial, eligible patients were younger than 65 years, with multiple myeloma that had not progressed in the interval between first-line ASCT, performed within the previous 6 months, and randomization, and who had normal liver function tests and blood cell counts.
In CALGB 100104, after undergoing ASCT, 460 patients were randomly assigned to lenalidomide (starting at a dose of 10 mg/day) or placebo between day 100 and day 110 after transplantation. In IFM 2005-02, after undergoing ASCT, 614 patients were randomized 1:1 to receive either consolidation treatment with lenalidomide (at a dose of 25 mg/day on days 1-21 of each 28-day cycle for 2 cycles) followed by maintenance with lenalidomide (10 mg/day for the first 3 months, increasing to 15 mg if tolerated), or the same consolidation treatment followed by maintenance therapy with placebo.
The primary endpoint of CALGB 100104 was time to progression (TTP) and lenalidomide was associated with a significantly longer TTP. Median PFS was also improved by around 15 months (hazard ratio [HR], 0.38; P < .001). In a more recent long-term PFS analysis, median PFS was 5.7 years in the lenalidomide arm compared with 1.9 years with placebo, a difference of 3.8 years (HR, 0.38).3
The primary endpoint for IFM 2005-02 was PFS and lenalidomide maintenance therapy resulted in a significant improvement in PFS in both the originally published study (18-month PFS advantage) and long-term follow-up. The most recent PFS analysis demonstrated a PFS of 3.9 years for lenalidomide, compared with 2 years for no maintenance, a difference of 1.9 years (HR, 0.53). Although the studies were not powered for an overall survival (OS) endpoint, a descriptive analysis showed a median OS of 9.3 years, compared with 7 years in CALGB 100104, and 8.8 years compared with 7.3 years in IFM 2005-02.
In a meta-analysis of data pooled from these 2 studies and a third randomized trial (GIMEMA-RVMM-PI-209),4 which was presented at the 2016 annual meeting of the American Society of Clinical Oncology, maintenance therapy with lenalidomide following frontline treatment with high-dose melphalan and ASCT reduced the risk of death by 26% compared with placebo or no maintenance therapy, prompting suggestions that lenalidomide become standard of care in this setting.
The safety profile of lenalidomide in this setting was similar to that previously described in other studies. The most frequently reported adverse events (AEs), across both studies, were neutropenia, thrombocytopenia, leukopenia, anemia, upper respiratory tract infection, bronchitis, nasopharyngitis, cough, gastroenteritis, diarrhea, rash, fatigue, asthenia, muscle spasm, and pyrexia. The most common grade 3/4 AEs included neutropenia, thrombocytopenia, and leukopenia. AEs were generally most common in the first 6 months of treatment and subsequently declined in frequency over time or remained stable.
The prescribing information carries warnings and precautions about embryo-fetal toxicity, hematologic toxicity, venous/arterial thromboembolic events, secondary primary malignancies, hepatotoxicity, allergic reactions, tumor lysis syndrome, and thyroid disorders.5 Given its teratogenic effects, lenalidomide is only available through a restricted program under a risk evaluation mitigation strategy.
Patients with neutropenia should be monitored for signs of infections, patients advised to look for signs of bleeding or bruising, and weekly complete blood count performed for the first 2 cycles, on days 1 and 15 of cycle 3 and every 4 weeks thereafter.
Action should be taken to try to reduce the risk of venous and arterial thromboembolic events where possible and thrombophylaxis is recommended, based on the assessment of the underlying risk. Since lenalidomide can increase the risk of secondary primary malignancies, each case should be evaluated for risk-to-benefit ratio.
Liver enzymes should be monitored periodically and treatment interrupted upon their elevation, resuming at a lower dose if levels return to baseline values. Patients who have a history of grade 4 rash following thalidomide treatment should not receive lenalidomide. If grade 2-3 skin rash occurs, treatment interruption or discontinuation should be considered and lenalidomide should be discontinued in the event of angioedema, grade 4 rash, exfoliative or bullous rash, or if Stevens-Johnson s
Patients with high tumor burden prior to treatment are at highest risk of tumor lysis syndrome and should be monitored closely and appropriate precautions taken, and thyroid function should be measured before and during lenalidomide treatment to address potential thyroid disorders. Lenalidomide is marketed as Revlimid by Celgene Corporation.
The treatment of multiple myeloma has been revolutionized in the past few decades, with the introduction of numerous novel drug classes that have more than doubled median survival times. The immunomodulatory drug (IMiD), lenalidomide, forms the backbone of the majority of treatment paradigms, first receiving US Food and Drug Administration approval in 2006 for use in combination with dexamethasone in previously treated patients with multiple myeloma. Since then, approved indications for lenalidomide in multiple myeloma have continued to expand.
Most recently, on February 22, 2017, lenalidomide was approved for use as maintenance therapy following autologous stem cell transplant (ASCT), making it the first and only treatment available in this setting. This approval was based on 2 randomized, controlled trials that evaluated the efficacy and safety of lenalidomide in more than 1,000 patients in this setting and demonstrated a significant advantage in progression-free survival (PFS) compared with patients receiving placebo.
CALGB 1001041 and IFM 2005-022 were randomized, double-blind phase 3 trials conducted at 47 locations across the United States and 78 centers in France, Belgium, and Switzerland, respectively. In the CALGB trial, eligible patients were 18-70 years of age, with a European Cooperative Oncology Group (ECOG) performance status of 0 or 1, symptomatic disease requiring treatment (Durie-Salmon stage ≥1), and who received any induction therapy of 2-12 months duration. In the IFM trial, eligible patients were younger than 65 years, with multiple myeloma that had not progressed in the interval between first-line ASCT, performed within the previous 6 months, and randomization, and who had normal liver function tests and blood cell counts.
In CALGB 100104, after undergoing ASCT, 460 patients were randomly assigned to lenalidomide (starting at a dose of 10 mg/day) or placebo between day 100 and day 110 after transplantation. In IFM 2005-02, after undergoing ASCT, 614 patients were randomized 1:1 to receive either consolidation treatment with lenalidomide (at a dose of 25 mg/day on days 1-21 of each 28-day cycle for 2 cycles) followed by maintenance with lenalidomide (10 mg/day for the first 3 months, increasing to 15 mg if tolerated), or the same consolidation treatment followed by maintenance therapy with placebo.
The primary endpoint of CALGB 100104 was time to progression (TTP) and lenalidomide was associated with a significantly longer TTP. Median PFS was also improved by around 15 months (hazard ratio [HR], 0.38; P < .001). In a more recent long-term PFS analysis, median PFS was 5.7 years in the lenalidomide arm compared with 1.9 years with placebo, a difference of 3.8 years (HR, 0.38).3
The primary endpoint for IFM 2005-02 was PFS and lenalidomide maintenance therapy resulted in a significant improvement in PFS in both the originally published study (18-month PFS advantage) and long-term follow-up. The most recent PFS analysis demonstrated a PFS of 3.9 years for lenalidomide, compared with 2 years for no maintenance, a difference of 1.9 years (HR, 0.53). Although the studies were not powered for an overall survival (OS) endpoint, a descriptive analysis showed a median OS of 9.3 years, compared with 7 years in CALGB 100104, and 8.8 years compared with 7.3 years in IFM 2005-02.
In a meta-analysis of data pooled from these 2 studies and a third randomized trial (GIMEMA-RVMM-PI-209),4 which was presented at the 2016 annual meeting of the American Society of Clinical Oncology, maintenance therapy with lenalidomide following frontline treatment with high-dose melphalan and ASCT reduced the risk of death by 26% compared with placebo or no maintenance therapy, prompting suggestions that lenalidomide become standard of care in this setting.
The safety profile of lenalidomide in this setting was similar to that previously described in other studies. The most frequently reported adverse events (AEs), across both studies, were neutropenia, thrombocytopenia, leukopenia, anemia, upper respiratory tract infection, bronchitis, nasopharyngitis, cough, gastroenteritis, diarrhea, rash, fatigue, asthenia, muscle spasm, and pyrexia. The most common grade 3/4 AEs included neutropenia, thrombocytopenia, and leukopenia. AEs were generally most common in the first 6 months of treatment and subsequently declined in frequency over time or remained stable.
The prescribing information carries warnings and precautions about embryo-fetal toxicity, hematologic toxicity, venous/arterial thromboembolic events, secondary primary malignancies, hepatotoxicity, allergic reactions, tumor lysis syndrome, and thyroid disorders.5 Given its teratogenic effects, lenalidomide is only available through a restricted program under a risk evaluation mitigation strategy.
Patients with neutropenia should be monitored for signs of infections, patients advised to look for signs of bleeding or bruising, and weekly complete blood count performed for the first 2 cycles, on days 1 and 15 of cycle 3 and every 4 weeks thereafter.
Action should be taken to try to reduce the risk of venous and arterial thromboembolic events where possible and thrombophylaxis is recommended, based on the assessment of the underlying risk. Since lenalidomide can increase the risk of secondary primary malignancies, each case should be evaluated for risk-to-benefit ratio.
Liver enzymes should be monitored periodically and treatment interrupted upon their elevation, resuming at a lower dose if levels return to baseline values. Patients who have a history of grade 4 rash following thalidomide treatment should not receive lenalidomide. If grade 2-3 skin rash occurs, treatment interruption or discontinuation should be considered and lenalidomide should be discontinued in the event of angioedema, grade 4 rash, exfoliative or bullous rash, or if Stevens-Johnson s
Patients with high tumor burden prior to treatment are at highest risk of tumor lysis syndrome and should be monitored closely and appropriate precautions taken, and thyroid function should be measured before and during lenalidomide treatment to address potential thyroid disorders. Lenalidomide is marketed as Revlimid by Celgene Corporation.
The treatment of multiple myeloma has been revolutionized in the past few decades, with the introduction of numerous novel drug classes that have more than doubled median survival times. The immunomodulatory drug (IMiD), lenalidomide, forms the backbone of the majority of treatment paradigms, first receiving US Food and Drug Administration approval in 2006 for use in combination with dexamethasone in previously treated patients with multiple myeloma. Since then, approved indications for lenalidomide in multiple myeloma have continued to expand.
Most recently, on February 22, 2017, lenalidomide was approved for use as maintenance therapy following autologous stem cell transplant (ASCT), making it the first and only treatment available in this setting. This approval was based on 2 randomized, controlled trials that evaluated the efficacy and safety of lenalidomide in more than 1,000 patients in this setting and demonstrated a significant advantage in progression-free survival (PFS) compared with patients receiving placebo.
CALGB 1001041 and IFM 2005-022 were randomized, double-blind phase 3 trials conducted at 47 locations across the United States and 78 centers in France, Belgium, and Switzerland, respectively. In the CALGB trial, eligible patients were 18-70 years of age, with a European Cooperative Oncology Group (ECOG) performance status of 0 or 1, symptomatic disease requiring treatment (Durie-Salmon stage ≥1), and who received any induction therapy of 2-12 months duration. In the IFM trial, eligible patients were younger than 65 years, with multiple myeloma that had not progressed in the interval between first-line ASCT, performed within the previous 6 months, and randomization, and who had normal liver function tests and blood cell counts.
In CALGB 100104, after undergoing ASCT, 460 patients were randomly assigned to lenalidomide (starting at a dose of 10 mg/day) or placebo between day 100 and day 110 after transplantation. In IFM 2005-02, after undergoing ASCT, 614 patients were randomized 1:1 to receive either consolidation treatment with lenalidomide (at a dose of 25 mg/day on days 1-21 of each 28-day cycle for 2 cycles) followed by maintenance with lenalidomide (10 mg/day for the first 3 months, increasing to 15 mg if tolerated), or the same consolidation treatment followed by maintenance therapy with placebo.
The primary endpoint of CALGB 100104 was time to progression (TTP) and lenalidomide was associated with a significantly longer TTP. Median PFS was also improved by around 15 months (hazard ratio [HR], 0.38; P < .001). In a more recent long-term PFS analysis, median PFS was 5.7 years in the lenalidomide arm compared with 1.9 years with placebo, a difference of 3.8 years (HR, 0.38).3
The primary endpoint for IFM 2005-02 was PFS and lenalidomide maintenance therapy resulted in a significant improvement in PFS in both the originally published study (18-month PFS advantage) and long-term follow-up. The most recent PFS analysis demonstrated a PFS of 3.9 years for lenalidomide, compared with 2 years for no maintenance, a difference of 1.9 years (HR, 0.53). Although the studies were not powered for an overall survival (OS) endpoint, a descriptive analysis showed a median OS of 9.3 years, compared with 7 years in CALGB 100104, and 8.8 years compared with 7.3 years in IFM 2005-02.
In a meta-analysis of data pooled from these 2 studies and a third randomized trial (GIMEMA-RVMM-PI-209),4 which was presented at the 2016 annual meeting of the American Society of Clinical Oncology, maintenance therapy with lenalidomide following frontline treatment with high-dose melphalan and ASCT reduced the risk of death by 26% compared with placebo or no maintenance therapy, prompting suggestions that lenalidomide become standard of care in this setting.
The safety profile of lenalidomide in this setting was similar to that previously described in other studies. The most frequently reported adverse events (AEs), across both studies, were neutropenia, thrombocytopenia, leukopenia, anemia, upper respiratory tract infection, bronchitis, nasopharyngitis, cough, gastroenteritis, diarrhea, rash, fatigue, asthenia, muscle spasm, and pyrexia. The most common grade 3/4 AEs included neutropenia, thrombocytopenia, and leukopenia. AEs were generally most common in the first 6 months of treatment and subsequently declined in frequency over time or remained stable.
The prescribing information carries warnings and precautions about embryo-fetal toxicity, hematologic toxicity, venous/arterial thromboembolic events, secondary primary malignancies, hepatotoxicity, allergic reactions, tumor lysis syndrome, and thyroid disorders.5 Given its teratogenic effects, lenalidomide is only available through a restricted program under a risk evaluation mitigation strategy.
Patients with neutropenia should be monitored for signs of infections, patients advised to look for signs of bleeding or bruising, and weekly complete blood count performed for the first 2 cycles, on days 1 and 15 of cycle 3 and every 4 weeks thereafter.
Action should be taken to try to reduce the risk of venous and arterial thromboembolic events where possible and thrombophylaxis is recommended, based on the assessment of the underlying risk. Since lenalidomide can increase the risk of secondary primary malignancies, each case should be evaluated for risk-to-benefit ratio.
Liver enzymes should be monitored periodically and treatment interrupted upon their elevation, resuming at a lower dose if levels return to baseline values. Patients who have a history of grade 4 rash following thalidomide treatment should not receive lenalidomide. If grade 2-3 skin rash occurs, treatment interruption or discontinuation should be considered and lenalidomide should be discontinued in the event of angioedema, grade 4 rash, exfoliative or bullous rash, or if Stevens-Johnson s
Patients with high tumor burden prior to treatment are at highest risk of tumor lysis syndrome and should be monitored closely and appropriate precautions taken, and thyroid function should be measured before and during lenalidomide treatment to address potential thyroid disorders. Lenalidomide is marketed as Revlimid by Celgene Corporation.
Trastuzumab-dkst approval adds to the biosimilar cancer drug market
The human epidermal growth factor receptor-2 (HER2)-targeting monoclonal antibody trastuzumab-dkst, was approved by the US Food and Drug Administration in 2017 for the treatment of patients with HER2-positive breast or metastatic gastric or gastroesophageal junction adenocarcinoma.1 Trastuzumab-dkst, marketed as Ogviri by Mylan NV and Biocon Ltd, is a copy, known as a biosimilar, of Genentech’s trastuzumab (Herceptin), which has been approved in the US since 1998. Genentech’s patent on trastuzumab expires in 2018
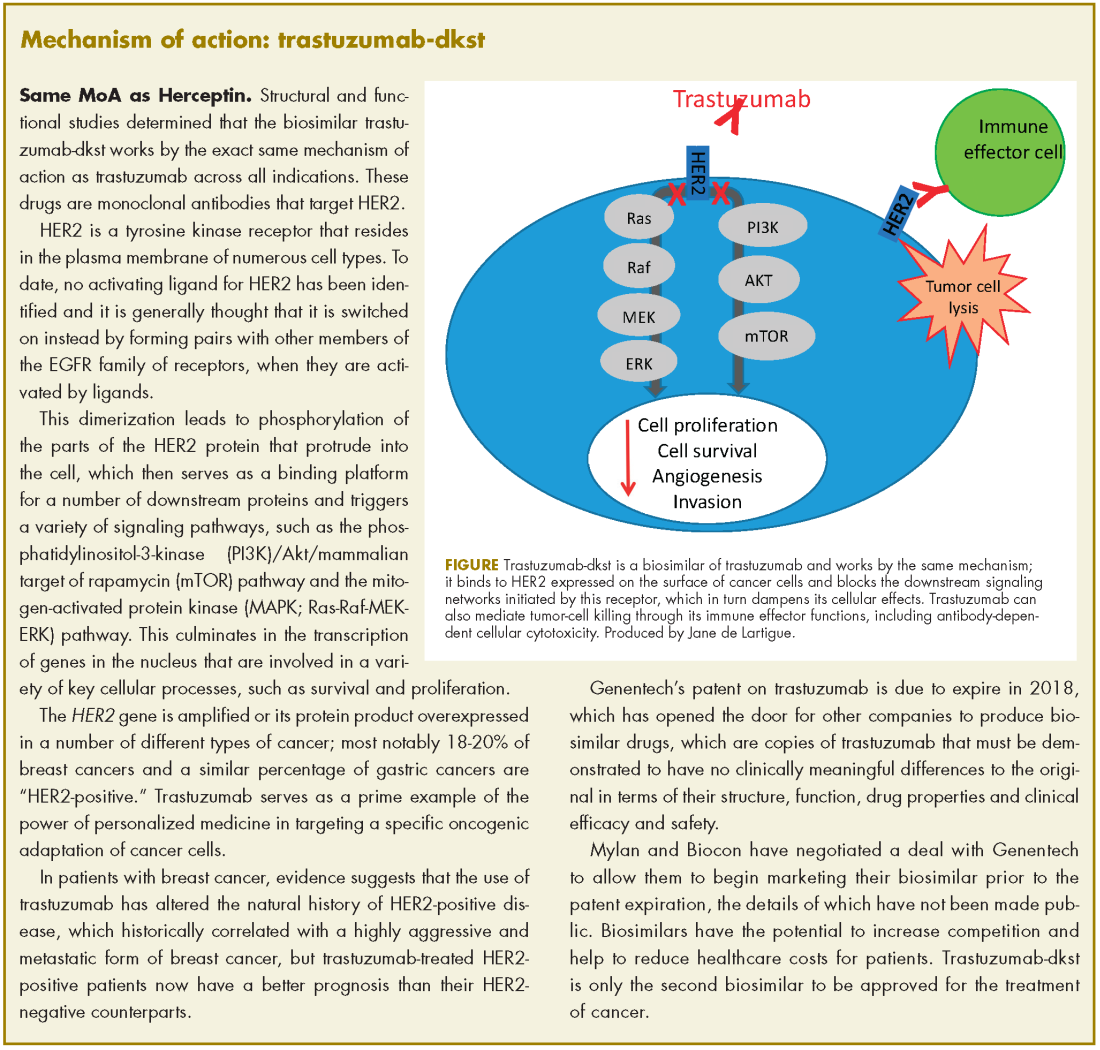
Approval was based on a comparison of the 2 drugs, which demonstrated that there were no clinically meaningful differences between the biosimilar and the reference product (trastuzumab) in terms of structure and function, pharmacokinetics (PKs), pharmacodynamics, and clinical efficacy and safety.
In structural and functional studies, trastuzumab-dkst was shown to have an identical amino acid sequence and a highly similar 3-dimensional structure, as well as equivalency in an inhibition of proliferation assay, a HER2-binding assay, and an antibody-dependent cellular cytotoxicity assay, compared with trastuzumab.
Two nonclinical animal studies were performed in cynomolgus monkeys; a single-dose comparative PK study and a 4-week, repeat-dose toxicity study. That was further supported by data from a single-dose, randomized, double-blind, comparative 3-way PK study (MYL-HER-1002) in which 120 healthy men were given an 8 mg/kg infusion of trastuzumab-dkst, US-approved trastuzumab, or European Union (EU)-approved trastuzumab.
The key clinical study was the phase 3 HERiTAge trial, a 2-part, multicenter, double-blind, randomized, parallel group trial that was performed in patients with HER2-positive metastatic breast cancer who had not been previously treated with either chemotherapy or trastuzumab in the metastatic setting.2
Eligible patients included males or females with measurably HER2-positive disease (as defined by HER2 overexpression determined by immunohistochemistry performed by a central laboratory), no exposure to chemotherapy or trastuzumab in the metastatic setting, an Eastern Cooperative Oncology Group Performance Status of 0 or 2, left ventricular ejection fraction (LVEF) within institutional range of normal, and who had completed adjuvant trastuzumab therapy at least 1 year before.
Patients with central nervous system metastases had to have stable disease after treatment, and hormonal agents were required to be discontinued before the start of the study. Patients with a history of unstable angina, heart failure, myocardial infarction less than 1 year from randomization, other clinically significant cardiac disease, grade 2 or higher peripheral neuropathy, a history of any other cancer within 4 years before screening, or any significant medical illness that increased treatment risk or impeded evaluation, were excluded from the study.
Patients were randomly assigned 1:1 to receive trastuzumab-dkst or trastuzumab, both in combination with paclitaxel or docetaxel, at a loading dose of 8 mg/kg, followed by a maintenance dose of 6 mg/kg, every 3 weeks for a minimum of 7 cycles in part 1 of the study. Patients who had stable disease or better were enrolled in part 2 and continued treatment until disease progression or unacceptable toxicity.
The primary endpoint was overall response rate (ORR) and, after 24 weeks, the ORR was 69.6% in the trastuzumab-dkst arm, compared with 64% in the trastuzumab arm, with a ratio of ORR of 1.09. Progression-free survival was also nearly identical in the 2 groups and median overall survival had not been reached in either arm.
The safety of the biosimilar and reference product were also highly similar. Serious adverse events occurred in 39.3%, compared with 37% of patients, respectively, with neutropenia the most frequently reported in both arms. Overall, treatment-emergent AEs occurred in 96.8%, compared with 94.7% of patients, respectively, with the majority of events mild or moderate in severity in both groups. This study also confirmed the low immunogenicity of the 2 drug products.
The prescribing information details the recommended doses of trastuzumab-dkst for each approved indication and warnings and precautions for cardiomyopathy, infusion reactions, pulmonary toxicity, exacerbation of chemotherapy-induced neutropenia and embryofetal toxicity.3
Patients should undergo thorough cardiac assessments, including baseline LVEF measurement immediately before starting therapy, every 3 months during therapy, and upon completion of therapy. Patients who complete adjuvant therapy should have cardiac assessments every 6 months for at least 2 years. Treatment should be withheld for ≥16% absolute decrease in LVEF from pre-treatment values or an LVEF value below institutional limits of normal and ≥10% absolute decrease in LVEF from pre-treatment values. When treatment is withheld for significant LVEF cardiac dysfunction, patients should undergo cardiac assessment at 4-week intervals.
To combat infusion reactions, infusion should be interrupted in all patients experiencing dyspnea or clinically significant hypotension and medical therapy administered. Patients should be evaluated and monitored carefully until signs and symptoms resolve and permanent discontinuation considered in patients with severe reactions. Patients should be warned of the potential for fetal harm with trastuzumab-dkst and of the need for effective contraceptive use during and for 6 months after treatment
1. FDA approves first biosimilar for the treatment of certain breast and stomach cancers. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm587378.htm. December 1, 2017. Accessed January 31, 2018.
2. Rugo HS, Barve A, Waller CF, et al. Effect of a proposed trastuzumab biosimilar compared with trastuzumab on overall response rate in patients with ERBB2 (HER2)-positive metastatic breast cancer: a randomized clinical trial. JAMA. 2017;317(1):37-47.
3. Ogviri (trastuzumab-dkst) injection, for intravenous use. Prescribing information. Mylan, GMBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761074s000lbl.pdf. December, 2017. Accessed July 31, 2015.
The human epidermal growth factor receptor-2 (HER2)-targeting monoclonal antibody trastuzumab-dkst, was approved by the US Food and Drug Administration in 2017 for the treatment of patients with HER2-positive breast or metastatic gastric or gastroesophageal junction adenocarcinoma.1 Trastuzumab-dkst, marketed as Ogviri by Mylan NV and Biocon Ltd, is a copy, known as a biosimilar, of Genentech’s trastuzumab (Herceptin), which has been approved in the US since 1998. Genentech’s patent on trastuzumab expires in 2018
Approval was based on a comparison of the 2 drugs, which demonstrated that there were no clinically meaningful differences between the biosimilar and the reference product (trastuzumab) in terms of structure and function, pharmacokinetics (PKs), pharmacodynamics, and clinical efficacy and safety.
In structural and functional studies, trastuzumab-dkst was shown to have an identical amino acid sequence and a highly similar 3-dimensional structure, as well as equivalency in an inhibition of proliferation assay, a HER2-binding assay, and an antibody-dependent cellular cytotoxicity assay, compared with trastuzumab.
Two nonclinical animal studies were performed in cynomolgus monkeys; a single-dose comparative PK study and a 4-week, repeat-dose toxicity study. That was further supported by data from a single-dose, randomized, double-blind, comparative 3-way PK study (MYL-HER-1002) in which 120 healthy men were given an 8 mg/kg infusion of trastuzumab-dkst, US-approved trastuzumab, or European Union (EU)-approved trastuzumab.
The key clinical study was the phase 3 HERiTAge trial, a 2-part, multicenter, double-blind, randomized, parallel group trial that was performed in patients with HER2-positive metastatic breast cancer who had not been previously treated with either chemotherapy or trastuzumab in the metastatic setting.2
Eligible patients included males or females with measurably HER2-positive disease (as defined by HER2 overexpression determined by immunohistochemistry performed by a central laboratory), no exposure to chemotherapy or trastuzumab in the metastatic setting, an Eastern Cooperative Oncology Group Performance Status of 0 or 2, left ventricular ejection fraction (LVEF) within institutional range of normal, and who had completed adjuvant trastuzumab therapy at least 1 year before.
Patients with central nervous system metastases had to have stable disease after treatment, and hormonal agents were required to be discontinued before the start of the study. Patients with a history of unstable angina, heart failure, myocardial infarction less than 1 year from randomization, other clinically significant cardiac disease, grade 2 or higher peripheral neuropathy, a history of any other cancer within 4 years before screening, or any significant medical illness that increased treatment risk or impeded evaluation, were excluded from the study.
Patients were randomly assigned 1:1 to receive trastuzumab-dkst or trastuzumab, both in combination with paclitaxel or docetaxel, at a loading dose of 8 mg/kg, followed by a maintenance dose of 6 mg/kg, every 3 weeks for a minimum of 7 cycles in part 1 of the study. Patients who had stable disease or better were enrolled in part 2 and continued treatment until disease progression or unacceptable toxicity.
The primary endpoint was overall response rate (ORR) and, after 24 weeks, the ORR was 69.6% in the trastuzumab-dkst arm, compared with 64% in the trastuzumab arm, with a ratio of ORR of 1.09. Progression-free survival was also nearly identical in the 2 groups and median overall survival had not been reached in either arm.
The safety of the biosimilar and reference product were also highly similar. Serious adverse events occurred in 39.3%, compared with 37% of patients, respectively, with neutropenia the most frequently reported in both arms. Overall, treatment-emergent AEs occurred in 96.8%, compared with 94.7% of patients, respectively, with the majority of events mild or moderate in severity in both groups. This study also confirmed the low immunogenicity of the 2 drug products.
The prescribing information details the recommended doses of trastuzumab-dkst for each approved indication and warnings and precautions for cardiomyopathy, infusion reactions, pulmonary toxicity, exacerbation of chemotherapy-induced neutropenia and embryofetal toxicity.3
Patients should undergo thorough cardiac assessments, including baseline LVEF measurement immediately before starting therapy, every 3 months during therapy, and upon completion of therapy. Patients who complete adjuvant therapy should have cardiac assessments every 6 months for at least 2 years. Treatment should be withheld for ≥16% absolute decrease in LVEF from pre-treatment values or an LVEF value below institutional limits of normal and ≥10% absolute decrease in LVEF from pre-treatment values. When treatment is withheld for significant LVEF cardiac dysfunction, patients should undergo cardiac assessment at 4-week intervals.
To combat infusion reactions, infusion should be interrupted in all patients experiencing dyspnea or clinically significant hypotension and medical therapy administered. Patients should be evaluated and monitored carefully until signs and symptoms resolve and permanent discontinuation considered in patients with severe reactions. Patients should be warned of the potential for fetal harm with trastuzumab-dkst and of the need for effective contraceptive use during and for 6 months after treatment
The human epidermal growth factor receptor-2 (HER2)-targeting monoclonal antibody trastuzumab-dkst, was approved by the US Food and Drug Administration in 2017 for the treatment of patients with HER2-positive breast or metastatic gastric or gastroesophageal junction adenocarcinoma.1 Trastuzumab-dkst, marketed as Ogviri by Mylan NV and Biocon Ltd, is a copy, known as a biosimilar, of Genentech’s trastuzumab (Herceptin), which has been approved in the US since 1998. Genentech’s patent on trastuzumab expires in 2018
Approval was based on a comparison of the 2 drugs, which demonstrated that there were no clinically meaningful differences between the biosimilar and the reference product (trastuzumab) in terms of structure and function, pharmacokinetics (PKs), pharmacodynamics, and clinical efficacy and safety.
In structural and functional studies, trastuzumab-dkst was shown to have an identical amino acid sequence and a highly similar 3-dimensional structure, as well as equivalency in an inhibition of proliferation assay, a HER2-binding assay, and an antibody-dependent cellular cytotoxicity assay, compared with trastuzumab.
Two nonclinical animal studies were performed in cynomolgus monkeys; a single-dose comparative PK study and a 4-week, repeat-dose toxicity study. That was further supported by data from a single-dose, randomized, double-blind, comparative 3-way PK study (MYL-HER-1002) in which 120 healthy men were given an 8 mg/kg infusion of trastuzumab-dkst, US-approved trastuzumab, or European Union (EU)-approved trastuzumab.
The key clinical study was the phase 3 HERiTAge trial, a 2-part, multicenter, double-blind, randomized, parallel group trial that was performed in patients with HER2-positive metastatic breast cancer who had not been previously treated with either chemotherapy or trastuzumab in the metastatic setting.2
Eligible patients included males or females with measurably HER2-positive disease (as defined by HER2 overexpression determined by immunohistochemistry performed by a central laboratory), no exposure to chemotherapy or trastuzumab in the metastatic setting, an Eastern Cooperative Oncology Group Performance Status of 0 or 2, left ventricular ejection fraction (LVEF) within institutional range of normal, and who had completed adjuvant trastuzumab therapy at least 1 year before.
Patients with central nervous system metastases had to have stable disease after treatment, and hormonal agents were required to be discontinued before the start of the study. Patients with a history of unstable angina, heart failure, myocardial infarction less than 1 year from randomization, other clinically significant cardiac disease, grade 2 or higher peripheral neuropathy, a history of any other cancer within 4 years before screening, or any significant medical illness that increased treatment risk or impeded evaluation, were excluded from the study.
Patients were randomly assigned 1:1 to receive trastuzumab-dkst or trastuzumab, both in combination with paclitaxel or docetaxel, at a loading dose of 8 mg/kg, followed by a maintenance dose of 6 mg/kg, every 3 weeks for a minimum of 7 cycles in part 1 of the study. Patients who had stable disease or better were enrolled in part 2 and continued treatment until disease progression or unacceptable toxicity.
The primary endpoint was overall response rate (ORR) and, after 24 weeks, the ORR was 69.6% in the trastuzumab-dkst arm, compared with 64% in the trastuzumab arm, with a ratio of ORR of 1.09. Progression-free survival was also nearly identical in the 2 groups and median overall survival had not been reached in either arm.
The safety of the biosimilar and reference product were also highly similar. Serious adverse events occurred in 39.3%, compared with 37% of patients, respectively, with neutropenia the most frequently reported in both arms. Overall, treatment-emergent AEs occurred in 96.8%, compared with 94.7% of patients, respectively, with the majority of events mild or moderate in severity in both groups. This study also confirmed the low immunogenicity of the 2 drug products.
The prescribing information details the recommended doses of trastuzumab-dkst for each approved indication and warnings and precautions for cardiomyopathy, infusion reactions, pulmonary toxicity, exacerbation of chemotherapy-induced neutropenia and embryofetal toxicity.3
Patients should undergo thorough cardiac assessments, including baseline LVEF measurement immediately before starting therapy, every 3 months during therapy, and upon completion of therapy. Patients who complete adjuvant therapy should have cardiac assessments every 6 months for at least 2 years. Treatment should be withheld for ≥16% absolute decrease in LVEF from pre-treatment values or an LVEF value below institutional limits of normal and ≥10% absolute decrease in LVEF from pre-treatment values. When treatment is withheld for significant LVEF cardiac dysfunction, patients should undergo cardiac assessment at 4-week intervals.
To combat infusion reactions, infusion should be interrupted in all patients experiencing dyspnea or clinically significant hypotension and medical therapy administered. Patients should be evaluated and monitored carefully until signs and symptoms resolve and permanent discontinuation considered in patients with severe reactions. Patients should be warned of the potential for fetal harm with trastuzumab-dkst and of the need for effective contraceptive use during and for 6 months after treatment
1. FDA approves first biosimilar for the treatment of certain breast and stomach cancers. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm587378.htm. December 1, 2017. Accessed January 31, 2018.
2. Rugo HS, Barve A, Waller CF, et al. Effect of a proposed trastuzumab biosimilar compared with trastuzumab on overall response rate in patients with ERBB2 (HER2)-positive metastatic breast cancer: a randomized clinical trial. JAMA. 2017;317(1):37-47.
3. Ogviri (trastuzumab-dkst) injection, for intravenous use. Prescribing information. Mylan, GMBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761074s000lbl.pdf. December, 2017. Accessed July 31, 2015.
1. FDA approves first biosimilar for the treatment of certain breast and stomach cancers. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm587378.htm. December 1, 2017. Accessed January 31, 2018.
2. Rugo HS, Barve A, Waller CF, et al. Effect of a proposed trastuzumab biosimilar compared with trastuzumab on overall response rate in patients with ERBB2 (HER2)-positive metastatic breast cancer: a randomized clinical trial. JAMA. 2017;317(1):37-47.
3. Ogviri (trastuzumab-dkst) injection, for intravenous use. Prescribing information. Mylan, GMBH. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761074s000lbl.pdf. December, 2017. Accessed July 31, 2015.
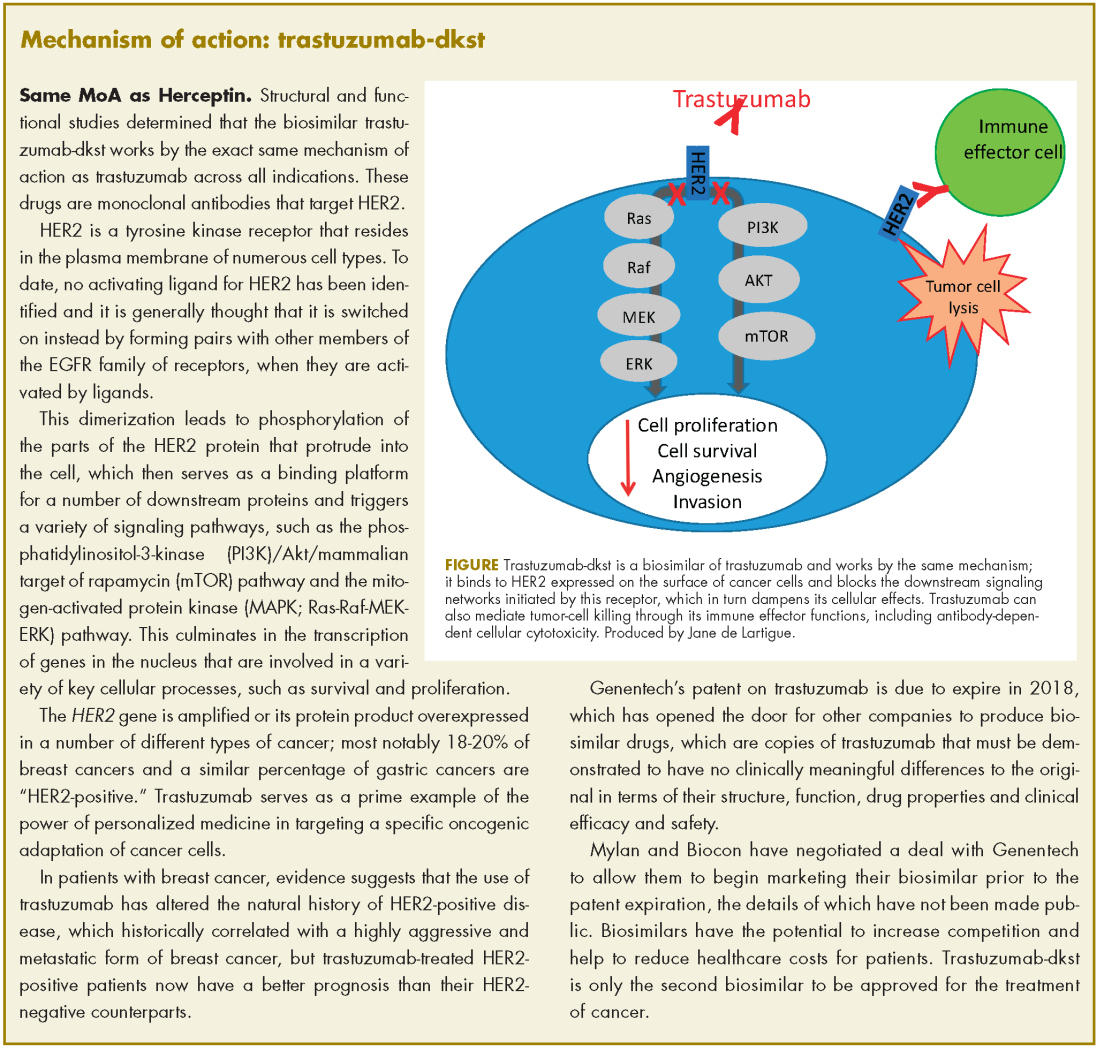
Pembrolizumab for dMMR/MSI-H tumors marks first tumor agnostic FDA approval
The United States Food and Drug Administration’s approval earlier this year of pembrolizumab marks the first tumor agnostic indication for a cancer drug.1,2 Accelerated approval was granted for the treatment of adult and pediatric patients with any unresectable or metastatic solid tumor that displays mismatch repair deficiencies (dMMR) or high levels of microsatellite instability (MSI-H) and who have progressed after previous treatment and have no satisfactory alternatives. It is also approved specifically for patients with MSI-H or dMMR colorectal cancer (CRC) that has progressed after treatment with a fluoropyrimidine, oxaliplatin, and irinotecan.
Pembrolizumab is a programmed cell death protein-1 (PD-1) receptor inhibitor that blocks the interaction between PD-1 and its ligand, PD-L1, restoring the activity of tumor-infiltrating T cells and boosting the anti-tumor immune response. It is thought to be particularly effective in dMMR/MSI-H tumors because they have a high mutational load and therefore display an abundance of antigens on their surfaces to provoke an immune response.
Approval for the drug was based on the demonstration of durable responses in 149 patients with MSI-H or dMMR cancers across 5 uncontrolled, multicohort, multicenter, single-arm trials. In all, 90 of the patients had CRC, and the remaining 59 patients had 1 of 14 other cancer types that included endometrial, biliary, gastric or gastroesophageal, pancreatic, and breast cancers.
Patients in these trials received pembrolizumab at 1 of 2 different doses, either 200 mg every 3 weeks or 10 mg/kg every 2 weeks, until unacceptable toxicity or disease progression that was symptomatic, rapidly progressive, required urgent intervention, or coincided with a decline in performance status. Treatment was administered for a maximum of 2 years. Patients with an active autoimmune disease or a medical condition that required immunosuppression were ineligible for treatment in all 5 studies.
The median age of enrolled patients was 55 years; 56% were men; 77% white, 19% Asian, 2% black; 98% had metastatic or unresectable disease; and all had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 (range, 0-5, where 0 denotes full activity and 1, restricted in physically strenuous activity but ambulatory). MSI-H and MMR status were identified prospectively using polymerase chain reaction and immunohistochemical analyses, respectively.
The primary endpoint was objective response rate (ORR), according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), as assessed by blinded independent central radiologist review, and response duration. The ORR across all five studies
The safety profile was consistent with previously reported safety data for pembrolizumab. The most common adverse events included fatigue, pruritus, diarrhea, decreased appetite, rash, pyrexia, cough, dyspnea, musculoskeletal pain, constipation, and nausea.
The prescribing information includes a “limitation of use” that states that pembroliumab’s safety and efficacy haven’t been established in pediatric patients with MSI-H cancers of the central nervous system.3 It also details warnings and precautions about immune-mediated toxicities, including pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, and renal dysfunction, among others.
Patients should be monitored for signs and symptoms of these toxicities and treated appropriately. Treatment should be withheld and corticosteroids should be administered for grade 2 or higher pneumonitis, colitis, hepatitis, and nephritis; and corticosteroids and hormone replacement as clinically indicated for endocrinopathies. It should also be withheld for aspartate aminotransferase (AST) or alanine aminotransferase (ALT) levels >3-5 times the upper limit of normal (ULN) or total bilirubin levels >1.5-3 times ULN.
Pembrolizumab should be permanently discontinued upon grade 3, 4, or recurrent grade 2 pneumonitis, colitis, nephritis/renal dysfunction, and endocrinopathies or for AST or ALT levels >5 times ULN or total bilirubin levels >3 times ULN. For patients with liver metastases who begin treatment with grade 2 AST or ALT, treatment should be permanently discontinued following increases of more than 50%, relative to baseline, that last for at least 1 week.
Health care providers should also bear in mind that pembrolizumab can, more rarely, cause other immune-mediated toxicities, such as arthritis and exfoliative rash that may require treatment and, based on its mechanism of action, pembrolizumab can also cause fetal harm. Patients with reproductive potential should be advised of the implications. Pembrolizumab is marketed as Keytruda by Merck & Co Inc.
1. United States Food and Drug Administration. FDA grants accelerated approval to pembrolizumab for tissue/site agnostic indication. US FDA Web site. https://www.fda.gov/drugs/informationondrugs/ approveddrugs/ucm560040.htm. Last updated May 30, 2017. Accessed July 15, 2017.
2. Merck. News Release. FDA Approves Merck’s KEYTRUDA (pembrolizumab) for Adult and Pediatric Patients with Unresectable or Metastatic, Microsatellite Instability-High (MSI-H) or Mismatch Repair De[1]cient (dMMR) Solid Tumors. http://www.mrknewsroom. com/news-release/prescription-medicine-news/fda-approvesmercks- keytruda-pembrolizumab-adult-and-pediatr. Last updated May 23, 2017. Accessed July 17, 2017.
3. Keytruda (pembrolizumab) for injection, for intravenous use. Prescribing information. Merck & Co Inc. https://www.merck.com/ product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf. Posted May 2017. Accessed July 15, 2017.
The United States Food and Drug Administration’s approval earlier this year of pembrolizumab marks the first tumor agnostic indication for a cancer drug.1,2 Accelerated approval was granted for the treatment of adult and pediatric patients with any unresectable or metastatic solid tumor that displays mismatch repair deficiencies (dMMR) or high levels of microsatellite instability (MSI-H) and who have progressed after previous treatment and have no satisfactory alternatives. It is also approved specifically for patients with MSI-H or dMMR colorectal cancer (CRC) that has progressed after treatment with a fluoropyrimidine, oxaliplatin, and irinotecan.
Pembrolizumab is a programmed cell death protein-1 (PD-1) receptor inhibitor that blocks the interaction between PD-1 and its ligand, PD-L1, restoring the activity of tumor-infiltrating T cells and boosting the anti-tumor immune response. It is thought to be particularly effective in dMMR/MSI-H tumors because they have a high mutational load and therefore display an abundance of antigens on their surfaces to provoke an immune response.
Approval for the drug was based on the demonstration of durable responses in 149 patients with MSI-H or dMMR cancers across 5 uncontrolled, multicohort, multicenter, single-arm trials. In all, 90 of the patients had CRC, and the remaining 59 patients had 1 of 14 other cancer types that included endometrial, biliary, gastric or gastroesophageal, pancreatic, and breast cancers.
Patients in these trials received pembrolizumab at 1 of 2 different doses, either 200 mg every 3 weeks or 10 mg/kg every 2 weeks, until unacceptable toxicity or disease progression that was symptomatic, rapidly progressive, required urgent intervention, or coincided with a decline in performance status. Treatment was administered for a maximum of 2 years. Patients with an active autoimmune disease or a medical condition that required immunosuppression were ineligible for treatment in all 5 studies.
The median age of enrolled patients was 55 years; 56% were men; 77% white, 19% Asian, 2% black; 98% had metastatic or unresectable disease; and all had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 (range, 0-5, where 0 denotes full activity and 1, restricted in physically strenuous activity but ambulatory). MSI-H and MMR status were identified prospectively using polymerase chain reaction and immunohistochemical analyses, respectively.
The primary endpoint was objective response rate (ORR), according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), as assessed by blinded independent central radiologist review, and response duration. The ORR across all five studies
The safety profile was consistent with previously reported safety data for pembrolizumab. The most common adverse events included fatigue, pruritus, diarrhea, decreased appetite, rash, pyrexia, cough, dyspnea, musculoskeletal pain, constipation, and nausea.
The prescribing information includes a “limitation of use” that states that pembroliumab’s safety and efficacy haven’t been established in pediatric patients with MSI-H cancers of the central nervous system.3 It also details warnings and precautions about immune-mediated toxicities, including pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, and renal dysfunction, among others.
Patients should be monitored for signs and symptoms of these toxicities and treated appropriately. Treatment should be withheld and corticosteroids should be administered for grade 2 or higher pneumonitis, colitis, hepatitis, and nephritis; and corticosteroids and hormone replacement as clinically indicated for endocrinopathies. It should also be withheld for aspartate aminotransferase (AST) or alanine aminotransferase (ALT) levels >3-5 times the upper limit of normal (ULN) or total bilirubin levels >1.5-3 times ULN.
Pembrolizumab should be permanently discontinued upon grade 3, 4, or recurrent grade 2 pneumonitis, colitis, nephritis/renal dysfunction, and endocrinopathies or for AST or ALT levels >5 times ULN or total bilirubin levels >3 times ULN. For patients with liver metastases who begin treatment with grade 2 AST or ALT, treatment should be permanently discontinued following increases of more than 50%, relative to baseline, that last for at least 1 week.
Health care providers should also bear in mind that pembrolizumab can, more rarely, cause other immune-mediated toxicities, such as arthritis and exfoliative rash that may require treatment and, based on its mechanism of action, pembrolizumab can also cause fetal harm. Patients with reproductive potential should be advised of the implications. Pembrolizumab is marketed as Keytruda by Merck & Co Inc.
The United States Food and Drug Administration’s approval earlier this year of pembrolizumab marks the first tumor agnostic indication for a cancer drug.1,2 Accelerated approval was granted for the treatment of adult and pediatric patients with any unresectable or metastatic solid tumor that displays mismatch repair deficiencies (dMMR) or high levels of microsatellite instability (MSI-H) and who have progressed after previous treatment and have no satisfactory alternatives. It is also approved specifically for patients with MSI-H or dMMR colorectal cancer (CRC) that has progressed after treatment with a fluoropyrimidine, oxaliplatin, and irinotecan.
Pembrolizumab is a programmed cell death protein-1 (PD-1) receptor inhibitor that blocks the interaction between PD-1 and its ligand, PD-L1, restoring the activity of tumor-infiltrating T cells and boosting the anti-tumor immune response. It is thought to be particularly effective in dMMR/MSI-H tumors because they have a high mutational load and therefore display an abundance of antigens on their surfaces to provoke an immune response.
Approval for the drug was based on the demonstration of durable responses in 149 patients with MSI-H or dMMR cancers across 5 uncontrolled, multicohort, multicenter, single-arm trials. In all, 90 of the patients had CRC, and the remaining 59 patients had 1 of 14 other cancer types that included endometrial, biliary, gastric or gastroesophageal, pancreatic, and breast cancers.
Patients in these trials received pembrolizumab at 1 of 2 different doses, either 200 mg every 3 weeks or 10 mg/kg every 2 weeks, until unacceptable toxicity or disease progression that was symptomatic, rapidly progressive, required urgent intervention, or coincided with a decline in performance status. Treatment was administered for a maximum of 2 years. Patients with an active autoimmune disease or a medical condition that required immunosuppression were ineligible for treatment in all 5 studies.
The median age of enrolled patients was 55 years; 56% were men; 77% white, 19% Asian, 2% black; 98% had metastatic or unresectable disease; and all had an Eastern Cooperative Oncology Group Performance Status of 0 or 1 (range, 0-5, where 0 denotes full activity and 1, restricted in physically strenuous activity but ambulatory). MSI-H and MMR status were identified prospectively using polymerase chain reaction and immunohistochemical analyses, respectively.
The primary endpoint was objective response rate (ORR), according to Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), as assessed by blinded independent central radiologist review, and response duration. The ORR across all five studies
The safety profile was consistent with previously reported safety data for pembrolizumab. The most common adverse events included fatigue, pruritus, diarrhea, decreased appetite, rash, pyrexia, cough, dyspnea, musculoskeletal pain, constipation, and nausea.
The prescribing information includes a “limitation of use” that states that pembroliumab’s safety and efficacy haven’t been established in pediatric patients with MSI-H cancers of the central nervous system.3 It also details warnings and precautions about immune-mediated toxicities, including pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, and renal dysfunction, among others.
Patients should be monitored for signs and symptoms of these toxicities and treated appropriately. Treatment should be withheld and corticosteroids should be administered for grade 2 or higher pneumonitis, colitis, hepatitis, and nephritis; and corticosteroids and hormone replacement as clinically indicated for endocrinopathies. It should also be withheld for aspartate aminotransferase (AST) or alanine aminotransferase (ALT) levels >3-5 times the upper limit of normal (ULN) or total bilirubin levels >1.5-3 times ULN.
Pembrolizumab should be permanently discontinued upon grade 3, 4, or recurrent grade 2 pneumonitis, colitis, nephritis/renal dysfunction, and endocrinopathies or for AST or ALT levels >5 times ULN or total bilirubin levels >3 times ULN. For patients with liver metastases who begin treatment with grade 2 AST or ALT, treatment should be permanently discontinued following increases of more than 50%, relative to baseline, that last for at least 1 week.
Health care providers should also bear in mind that pembrolizumab can, more rarely, cause other immune-mediated toxicities, such as arthritis and exfoliative rash that may require treatment and, based on its mechanism of action, pembrolizumab can also cause fetal harm. Patients with reproductive potential should be advised of the implications. Pembrolizumab is marketed as Keytruda by Merck & Co Inc.
1. United States Food and Drug Administration. FDA grants accelerated approval to pembrolizumab for tissue/site agnostic indication. US FDA Web site. https://www.fda.gov/drugs/informationondrugs/ approveddrugs/ucm560040.htm. Last updated May 30, 2017. Accessed July 15, 2017.
2. Merck. News Release. FDA Approves Merck’s KEYTRUDA (pembrolizumab) for Adult and Pediatric Patients with Unresectable or Metastatic, Microsatellite Instability-High (MSI-H) or Mismatch Repair De[1]cient (dMMR) Solid Tumors. http://www.mrknewsroom. com/news-release/prescription-medicine-news/fda-approvesmercks- keytruda-pembrolizumab-adult-and-pediatr. Last updated May 23, 2017. Accessed July 17, 2017.
3. Keytruda (pembrolizumab) for injection, for intravenous use. Prescribing information. Merck & Co Inc. https://www.merck.com/ product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf. Posted May 2017. Accessed July 15, 2017.
1. United States Food and Drug Administration. FDA grants accelerated approval to pembrolizumab for tissue/site agnostic indication. US FDA Web site. https://www.fda.gov/drugs/informationondrugs/ approveddrugs/ucm560040.htm. Last updated May 30, 2017. Accessed July 15, 2017.
2. Merck. News Release. FDA Approves Merck’s KEYTRUDA (pembrolizumab) for Adult and Pediatric Patients with Unresectable or Metastatic, Microsatellite Instability-High (MSI-H) or Mismatch Repair De[1]cient (dMMR) Solid Tumors. http://www.mrknewsroom. com/news-release/prescription-medicine-news/fda-approvesmercks- keytruda-pembrolizumab-adult-and-pediatr. Last updated May 23, 2017. Accessed July 17, 2017.
3. Keytruda (pembrolizumab) for injection, for intravenous use. Prescribing information. Merck & Co Inc. https://www.merck.com/ product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf. Posted May 2017. Accessed July 15, 2017.

Brigatinib approval yields additional treatment options for crizotinib-resistant, ALK-positive NSCLC patients
The accelerated approval by the United States Food and Drug Administration (FDA) of the anaplastic lymphoma kinase (ALK) inhibitor brigatinib, marked the fourth approved drug in this class.1 The most recent approval expands the available treatment options for patients with metastatic ALK-positive non–small-cell lung cancer (NSCLC) whose disease is no longer responding to the first-line ALK inhibitor crizotinib. The FDA based its decision on the results of the phase 2 ALTA trial, in which a significant proportion of patients experienced tumor shrinkage.2
The pivotal trial was a noncomparative, 2-arm, open-label, multicenter study that was carried out during June 2014-September 2015 at 71 centers across 18 countries. Eligible patients were 18 years or older, with locally advanced or metastatic ALK-positive NSCLC, disease progression while taking crizotinib, at least 1 measurable lesion, adequate organ and hematologic function, and Eastern Cooperative Oncology Group (ECOG) performance status of ≤2 (range, 0-5, where 0 means the patient is fully active, and 2, ambulatory and capable of all self-care but not able to carry out any work activities).
Patients were excluded from the trial if they had received previous ALK inhibitor therapy, other than crizotinib, or had received crizotinib within 3 days of the first dose of brigatinib, or they had received chemotherapy, radiation therapy, or investigational drugs within 14 days or monoclonal antibody therapy within 30 days of the first dose of the study drug. Anyone with a history or the presence of pulmonary interstitial disease or drug-related pneumonitis or symptomatic central nervous system (CNS) metastases that were neurologically unstable or required an increasing dose of corticosteroids was also ineligible.
A total of 222 patients were randomized to receive one of two brigatinib doses, either 90 mg daily or 180 mg daily after a 7-day lead-in at 90 mg (the latter to help mitigate pulmonary adverse events observed in previous studies). Randomization was stratified according to baseline brain metastases (present or absent) and best investigator-assessed response to crizotinib (complete response [CR] or partial response [PR] vs other or unknown)
Chest and abdomen imaging by computed-tomography (CT) or magnetic resonance imaging (MRI) with contrast were performed to assess disease at screening and every 8 weeks through cycle 15, and then every 12 weeks until disease progression. Contrast-enhanced brain MRI was carried out at screening and repeated after baseline for the 68% of patients who had CNS metastases at the time of enrollment.
The primary endpoint was confirmed investigator-assessed objective response rate (ORR) per Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), and secondary endpoints included CNS response, duration of response (DoR), progression-free and overall survival (PFS and OS, respectively). ORRs for the 90-mg and 180-mg doses were 48% and 53%, respectively. Responses occurred quickly and were durable in both arms; after a median follow-up of 8 months, median DoR was 13.8 months for both doses. Among the patients with brain metastases, the intracranial response rates for the two doses were 42% and 67%, respectively, notable because of the poor ability of crizotinib to penetrate the blood-brain barrier.
Other secondary outcomes also favored the 180-mg dose. Investigator-assessed PFS for the 90-mg and 180-mg doses were 9.2 months and 12.9 months, respectively, and estimated 1-year OS was 71% and 80%, respectively, the latter representing a nonstatistically significant 43% reduction in the risk of death with the 180 mg dose. There were 4 confirmed CRs in the 180-mg arm and 1 in the 90-mg arm.
The safety of brigatinib was evaluated in 219 patients who received at least 1 dose of brigatinib. Treatment was discontinued in 8% of patients in the 180-mg arm and 3% in the 90-mg arm because of adverse events (AEs). The most common AEs were nausea, diarrhea, fatigue, cough, and headache, and visual disturbances also occurred. The most common serious AEs were pneumonia and interstitial lung disease/pneumonitis.
The prescribing information details warnings and precautions about these and other potential toxicities, including hypertension, bradycardia, creatine phosphokinase (CPK) and pancreatic enzyme elevation, and hyperglycemia.3 Patients should be monitored for new or worsening respiratory symptoms, especially during the first week of initiating brigatinib treatment; blood pressure should be controlled before treatment initiation and monitored after 2 weeks and at least monthly thereafter; heart rate and blood pressure should be monitored frequently; patients should be advised to report any visual symptoms, or any unexplained muscle pain, tenderness or weakness; CPK, lipase, and amylase levels should be monitored during treatment, and fasting glucose tested before starting treatment and periodically thereafter.
Brigatinib should be withheld in any patient with new or worsening respiratory symptoms, for grade 3 hypertension despite optimal antihypertensive therapy, for symptomatic bradycardia, for patients with new or worsening visual symptoms of grade 2 or above, for grade 3 or 4 CPK or pancreatic enzyme elevation, or if adequate hyperglycemia control cannot be achieved. Treatment should be permanently discontinued for grade 3 or 4 or recurrent interstitial lung disease/pneumonitis, grade 4 or recurrent grade 3 hypertension, life-threatening bradycardia, and grade 4 visual disturbance.
Based on its mechanism of action, brigatinib can cause fetal harm and patients of reproductive potential should be advised of the risks and necessary precautions. Brigatinib is marketed as Alunbrig. It was discovered by Ariad Pharmaceuticals Inc, which was acquired by Takeda in February 2017.
1. United States Food and Drug Administration. Brigatinib. US FDA Web site. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm555841.htm. Last updated April 28, 2017. Accessed July 15, 2017
2. Kim D-W, Tiseo M, Ahn M-J, Reckamp KL, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non–small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol. 2017;35(22):2490-2498.
3. Alunbrig (brigatinib) tablets, for oral use. Prescribing information. Ariad Pharmaceuticals Inc. https://www.alunbrig.com/assets/pi.pdf. Posted April 2017. Accessed July 15, 2017.
The accelerated approval by the United States Food and Drug Administration (FDA) of the anaplastic lymphoma kinase (ALK) inhibitor brigatinib, marked the fourth approved drug in this class.1 The most recent approval expands the available treatment options for patients with metastatic ALK-positive non–small-cell lung cancer (NSCLC) whose disease is no longer responding to the first-line ALK inhibitor crizotinib. The FDA based its decision on the results of the phase 2 ALTA trial, in which a significant proportion of patients experienced tumor shrinkage.2
The pivotal trial was a noncomparative, 2-arm, open-label, multicenter study that was carried out during June 2014-September 2015 at 71 centers across 18 countries. Eligible patients were 18 years or older, with locally advanced or metastatic ALK-positive NSCLC, disease progression while taking crizotinib, at least 1 measurable lesion, adequate organ and hematologic function, and Eastern Cooperative Oncology Group (ECOG) performance status of ≤2 (range, 0-5, where 0 means the patient is fully active, and 2, ambulatory and capable of all self-care but not able to carry out any work activities).
Patients were excluded from the trial if they had received previous ALK inhibitor therapy, other than crizotinib, or had received crizotinib within 3 days of the first dose of brigatinib, or they had received chemotherapy, radiation therapy, or investigational drugs within 14 days or monoclonal antibody therapy within 30 days of the first dose of the study drug. Anyone with a history or the presence of pulmonary interstitial disease or drug-related pneumonitis or symptomatic central nervous system (CNS) metastases that were neurologically unstable or required an increasing dose of corticosteroids was also ineligible.
A total of 222 patients were randomized to receive one of two brigatinib doses, either 90 mg daily or 180 mg daily after a 7-day lead-in at 90 mg (the latter to help mitigate pulmonary adverse events observed in previous studies). Randomization was stratified according to baseline brain metastases (present or absent) and best investigator-assessed response to crizotinib (complete response [CR] or partial response [PR] vs other or unknown)
Chest and abdomen imaging by computed-tomography (CT) or magnetic resonance imaging (MRI) with contrast were performed to assess disease at screening and every 8 weeks through cycle 15, and then every 12 weeks until disease progression. Contrast-enhanced brain MRI was carried out at screening and repeated after baseline for the 68% of patients who had CNS metastases at the time of enrollment.
The primary endpoint was confirmed investigator-assessed objective response rate (ORR) per Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), and secondary endpoints included CNS response, duration of response (DoR), progression-free and overall survival (PFS and OS, respectively). ORRs for the 90-mg and 180-mg doses were 48% and 53%, respectively. Responses occurred quickly and were durable in both arms; after a median follow-up of 8 months, median DoR was 13.8 months for both doses. Among the patients with brain metastases, the intracranial response rates for the two doses were 42% and 67%, respectively, notable because of the poor ability of crizotinib to penetrate the blood-brain barrier.
Other secondary outcomes also favored the 180-mg dose. Investigator-assessed PFS for the 90-mg and 180-mg doses were 9.2 months and 12.9 months, respectively, and estimated 1-year OS was 71% and 80%, respectively, the latter representing a nonstatistically significant 43% reduction in the risk of death with the 180 mg dose. There were 4 confirmed CRs in the 180-mg arm and 1 in the 90-mg arm.
The safety of brigatinib was evaluated in 219 patients who received at least 1 dose of brigatinib. Treatment was discontinued in 8% of patients in the 180-mg arm and 3% in the 90-mg arm because of adverse events (AEs). The most common AEs were nausea, diarrhea, fatigue, cough, and headache, and visual disturbances also occurred. The most common serious AEs were pneumonia and interstitial lung disease/pneumonitis.
The prescribing information details warnings and precautions about these and other potential toxicities, including hypertension, bradycardia, creatine phosphokinase (CPK) and pancreatic enzyme elevation, and hyperglycemia.3 Patients should be monitored for new or worsening respiratory symptoms, especially during the first week of initiating brigatinib treatment; blood pressure should be controlled before treatment initiation and monitored after 2 weeks and at least monthly thereafter; heart rate and blood pressure should be monitored frequently; patients should be advised to report any visual symptoms, or any unexplained muscle pain, tenderness or weakness; CPK, lipase, and amylase levels should be monitored during treatment, and fasting glucose tested before starting treatment and periodically thereafter.
Brigatinib should be withheld in any patient with new or worsening respiratory symptoms, for grade 3 hypertension despite optimal antihypertensive therapy, for symptomatic bradycardia, for patients with new or worsening visual symptoms of grade 2 or above, for grade 3 or 4 CPK or pancreatic enzyme elevation, or if adequate hyperglycemia control cannot be achieved. Treatment should be permanently discontinued for grade 3 or 4 or recurrent interstitial lung disease/pneumonitis, grade 4 or recurrent grade 3 hypertension, life-threatening bradycardia, and grade 4 visual disturbance.
Based on its mechanism of action, brigatinib can cause fetal harm and patients of reproductive potential should be advised of the risks and necessary precautions. Brigatinib is marketed as Alunbrig. It was discovered by Ariad Pharmaceuticals Inc, which was acquired by Takeda in February 2017.
The accelerated approval by the United States Food and Drug Administration (FDA) of the anaplastic lymphoma kinase (ALK) inhibitor brigatinib, marked the fourth approved drug in this class.1 The most recent approval expands the available treatment options for patients with metastatic ALK-positive non–small-cell lung cancer (NSCLC) whose disease is no longer responding to the first-line ALK inhibitor crizotinib. The FDA based its decision on the results of the phase 2 ALTA trial, in which a significant proportion of patients experienced tumor shrinkage.2
The pivotal trial was a noncomparative, 2-arm, open-label, multicenter study that was carried out during June 2014-September 2015 at 71 centers across 18 countries. Eligible patients were 18 years or older, with locally advanced or metastatic ALK-positive NSCLC, disease progression while taking crizotinib, at least 1 measurable lesion, adequate organ and hematologic function, and Eastern Cooperative Oncology Group (ECOG) performance status of ≤2 (range, 0-5, where 0 means the patient is fully active, and 2, ambulatory and capable of all self-care but not able to carry out any work activities).
Patients were excluded from the trial if they had received previous ALK inhibitor therapy, other than crizotinib, or had received crizotinib within 3 days of the first dose of brigatinib, or they had received chemotherapy, radiation therapy, or investigational drugs within 14 days or monoclonal antibody therapy within 30 days of the first dose of the study drug. Anyone with a history or the presence of pulmonary interstitial disease or drug-related pneumonitis or symptomatic central nervous system (CNS) metastases that were neurologically unstable or required an increasing dose of corticosteroids was also ineligible.
A total of 222 patients were randomized to receive one of two brigatinib doses, either 90 mg daily or 180 mg daily after a 7-day lead-in at 90 mg (the latter to help mitigate pulmonary adverse events observed in previous studies). Randomization was stratified according to baseline brain metastases (present or absent) and best investigator-assessed response to crizotinib (complete response [CR] or partial response [PR] vs other or unknown)
Chest and abdomen imaging by computed-tomography (CT) or magnetic resonance imaging (MRI) with contrast were performed to assess disease at screening and every 8 weeks through cycle 15, and then every 12 weeks until disease progression. Contrast-enhanced brain MRI was carried out at screening and repeated after baseline for the 68% of patients who had CNS metastases at the time of enrollment.
The primary endpoint was confirmed investigator-assessed objective response rate (ORR) per Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), and secondary endpoints included CNS response, duration of response (DoR), progression-free and overall survival (PFS and OS, respectively). ORRs for the 90-mg and 180-mg doses were 48% and 53%, respectively. Responses occurred quickly and were durable in both arms; after a median follow-up of 8 months, median DoR was 13.8 months for both doses. Among the patients with brain metastases, the intracranial response rates for the two doses were 42% and 67%, respectively, notable because of the poor ability of crizotinib to penetrate the blood-brain barrier.
Other secondary outcomes also favored the 180-mg dose. Investigator-assessed PFS for the 90-mg and 180-mg doses were 9.2 months and 12.9 months, respectively, and estimated 1-year OS was 71% and 80%, respectively, the latter representing a nonstatistically significant 43% reduction in the risk of death with the 180 mg dose. There were 4 confirmed CRs in the 180-mg arm and 1 in the 90-mg arm.
The safety of brigatinib was evaluated in 219 patients who received at least 1 dose of brigatinib. Treatment was discontinued in 8% of patients in the 180-mg arm and 3% in the 90-mg arm because of adverse events (AEs). The most common AEs were nausea, diarrhea, fatigue, cough, and headache, and visual disturbances also occurred. The most common serious AEs were pneumonia and interstitial lung disease/pneumonitis.
The prescribing information details warnings and precautions about these and other potential toxicities, including hypertension, bradycardia, creatine phosphokinase (CPK) and pancreatic enzyme elevation, and hyperglycemia.3 Patients should be monitored for new or worsening respiratory symptoms, especially during the first week of initiating brigatinib treatment; blood pressure should be controlled before treatment initiation and monitored after 2 weeks and at least monthly thereafter; heart rate and blood pressure should be monitored frequently; patients should be advised to report any visual symptoms, or any unexplained muscle pain, tenderness or weakness; CPK, lipase, and amylase levels should be monitored during treatment, and fasting glucose tested before starting treatment and periodically thereafter.
Brigatinib should be withheld in any patient with new or worsening respiratory symptoms, for grade 3 hypertension despite optimal antihypertensive therapy, for symptomatic bradycardia, for patients with new or worsening visual symptoms of grade 2 or above, for grade 3 or 4 CPK or pancreatic enzyme elevation, or if adequate hyperglycemia control cannot be achieved. Treatment should be permanently discontinued for grade 3 or 4 or recurrent interstitial lung disease/pneumonitis, grade 4 or recurrent grade 3 hypertension, life-threatening bradycardia, and grade 4 visual disturbance.
Based on its mechanism of action, brigatinib can cause fetal harm and patients of reproductive potential should be advised of the risks and necessary precautions. Brigatinib is marketed as Alunbrig. It was discovered by Ariad Pharmaceuticals Inc, which was acquired by Takeda in February 2017.
1. United States Food and Drug Administration. Brigatinib. US FDA Web site. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm555841.htm. Last updated April 28, 2017. Accessed July 15, 2017
2. Kim D-W, Tiseo M, Ahn M-J, Reckamp KL, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non–small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol. 2017;35(22):2490-2498.
3. Alunbrig (brigatinib) tablets, for oral use. Prescribing information. Ariad Pharmaceuticals Inc. https://www.alunbrig.com/assets/pi.pdf. Posted April 2017. Accessed July 15, 2017.
1. United States Food and Drug Administration. Brigatinib. US FDA Web site. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm555841.htm. Last updated April 28, 2017. Accessed July 15, 2017
2. Kim D-W, Tiseo M, Ahn M-J, Reckamp KL, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non–small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol. 2017;35(22):2490-2498.
3. Alunbrig (brigatinib) tablets, for oral use. Prescribing information. Ariad Pharmaceuticals Inc. https://www.alunbrig.com/assets/pi.pdf. Posted April 2017. Accessed July 15, 2017.
PD-L1-targeting drug atezolizumab nabs approval for non-small cell lung cancer
The approval last fall by the US Food and Drug Administration (FDA) of the immune checkpoint inhibitor atezolizumab for the treatment of metastatic non–small cell lung cancer (NSCLC) marked the second approved indication for the drug in a single year. Atezolizumab, which targets the programmed cell death protein-ligand 1 (PD-L1), has a unique mechanism of action compared with other approved immune checkpoint inhibitors and had been previously approved for the treatment of patients with advanced urothelial carcinoma. The current approval was based on 2 international, randomized, open-label trials, involving more than 1,000 patients, in which atezolizumab outperformed the chemotherapeutic drug docetaxel.
The POPLAR trial1 was a phase 2 study performed at 61 academic centers and community oncology practices across 13 countries in Europe and North America and enrolled 287 patients, while the phase 3 OAK trial2 was carried out at 193 centers in 31 countries and enrolled 850 patients.
Eligibility criteria for the 2 studies were similar; patients were 18 years or older, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, had measurable disease as per Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), and adequate hematologic and end-organ function. Patients with asymptomatic or treated central nervous system (CNS) metastases were also eligible.
Patients were excluded from both studies if they had a history of autoimmune disease, pneumonitis or chronic viral diseases, or had received previous treatment with docetaxel, CD137 agonists, anti-CTLA4, anti-PD-L1 or anti-PD-1 therapeutics.
Patients were randomized 1:1 to receive atezolizumab, administered intravenously at a fixed dose of 1,200 mg, or a 75 mg/m2 dose of docetaxel, every 3 weeks on day 1 of each 3-week cycle. Randomization was stratified according to PD-L1 expression, number of previous chemotherapy regimens, and histology (squamous vs nonsquamous). Tumors were assessed at baseline and every 6 weeks for 36 weeks, and every 9 weeks thereafter until progression or, in patients who continued beyond progression, until discontinuation. PD-L1 expression was assessed prospectively on tumor cells and tumor-infiltrating immune cells, using the FDA-approved companion diagnostic, Ventana SP142 PD-L1 immunohistochemistry assay.
The primary endpoint in both studies was overall survival (OS), which was significantly improved in the atezolizumab treatment arm. In the POPLAR trial, over a minimum follow-up of 13 months, the median OS was 12.6 months in atezolizumab-treated patients, compared with 9.7 months in docetaxel-treated patients (hazard ratio [HR], 0.73; P = .04) and in the OAK trial, over a median follow-up of 21 months, the median OS was 15.7 months and 10.3 months, respectively.
The main difference between the 2 trials was that the OS benefit in the POPLAR trial reached statistical significance only in patients with the highest levels of PD-L1 expression, whereas in the OAK study, the OS was significantly improved regardless of PD-L1 expression status, although the greatest benefit was derived in patients with higher levels of PD-L1 expression (20.5 months and 8.9 months, respectively). The OS benefit was observed across all other prespecified subgroups, except in patients with EGFR mutation-positivity in the OAK trial. Progression-free survival and overall response rates were similar in the 2 treatment arms in both studies.
An additional 375 patients were enrolled in the OAK trial and included in the safety analyses. In both studies, the safety profile of atezolizumab was similar to that observed in previous studies of this drug. The most commonly observed adverse events (AEs) with atezolizumab treatment were fatigue, decreased appetite, dyspnea, cough, nausea, musculoskeletal pain, and constipation. Despite longer treatment duration, there were fewer incidences of grade 3/4 AEs with atezolizumab compared with docetaxel (37% vs 54%, respectively, in the OAK trial), a lower rate of discontinuation with atezolizumab treatment, and no deaths related to the study drug. The most common grade 3/4 AEs included dyspnea, pneumonia, hypoxia, hyponatremia, fatigue, and anemia. Clinically significant immune-related AEs included pneumonitis, hepatitis, colitis, and thyroid disease.
Atezolizumab is marketed as Tecentriq by Genentech and the prescribing information details warnings and precautions about immune-related AEs; pneumonitis, colitis, endocrinopathies (eg, thyroid disorders, adrenal insufficiency, and diabetes mellitus), and others, as well as infections, infusion-related reactions, and embryofetal toxicity.3
Atezolizumab treatment should be withheld for grade 2 pneumonitis; aspartate aminotransferase (AST) or alanine aminotransferase (ALT) levels of >3-5 times the upper limit of normal (ULN) or total bilirubin levels of >1.5 and up to 3 times the ULN; grade 2/3 diarrhea or colitis, adrenal insufficiency, hypothyroidism, or grade 3/4 hyperglycemia; grade 2 ocular inflammatory toxicity; grade 2/3 pancreatitis or grade 3/4 increases in amylase or lipase levels; grade 3/4 infection; grade 2 infusion-related reactions; and grade 3 rash. Treatment can then be resumed in patients whose AEs recover to grade 0 or 1.
Treatment should be permanently discontinued in the event of grade 3/4 pneumonitis; AST/ALT levels of >5 times the ULN or bilirubin levels of >3 times the ULN; grade 4 diarrhea or colitis; myasthenic syndrome/myasthenia gravis, Guillain-Barre syndrome or meningoencephalitis (all grades); grade 3/4 ocular inflammatory toxicity; grade 4 or recurrent (any grade) pancreatitis; grade 3/4 infusion-related reactions; or grade 4 rash. Patients should also be advised of the risk of fetal harm.
The approval last fall by the US Food and Drug Administration (FDA) of the immune checkpoint inhibitor atezolizumab for the treatment of metastatic non–small cell lung cancer (NSCLC) marked the second approved indication for the drug in a single year. Atezolizumab, which targets the programmed cell death protein-ligand 1 (PD-L1), has a unique mechanism of action compared with other approved immune checkpoint inhibitors and had been previously approved for the treatment of patients with advanced urothelial carcinoma. The current approval was based on 2 international, randomized, open-label trials, involving more than 1,000 patients, in which atezolizumab outperformed the chemotherapeutic drug docetaxel.
The POPLAR trial1 was a phase 2 study performed at 61 academic centers and community oncology practices across 13 countries in Europe and North America and enrolled 287 patients, while the phase 3 OAK trial2 was carried out at 193 centers in 31 countries and enrolled 850 patients.
Eligibility criteria for the 2 studies were similar; patients were 18 years or older, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, had measurable disease as per Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), and adequate hematologic and end-organ function. Patients with asymptomatic or treated central nervous system (CNS) metastases were also eligible.
Patients were excluded from both studies if they had a history of autoimmune disease, pneumonitis or chronic viral diseases, or had received previous treatment with docetaxel, CD137 agonists, anti-CTLA4, anti-PD-L1 or anti-PD-1 therapeutics.
Patients were randomized 1:1 to receive atezolizumab, administered intravenously at a fixed dose of 1,200 mg, or a 75 mg/m2 dose of docetaxel, every 3 weeks on day 1 of each 3-week cycle. Randomization was stratified according to PD-L1 expression, number of previous chemotherapy regimens, and histology (squamous vs nonsquamous). Tumors were assessed at baseline and every 6 weeks for 36 weeks, and every 9 weeks thereafter until progression or, in patients who continued beyond progression, until discontinuation. PD-L1 expression was assessed prospectively on tumor cells and tumor-infiltrating immune cells, using the FDA-approved companion diagnostic, Ventana SP142 PD-L1 immunohistochemistry assay.
The primary endpoint in both studies was overall survival (OS), which was significantly improved in the atezolizumab treatment arm. In the POPLAR trial, over a minimum follow-up of 13 months, the median OS was 12.6 months in atezolizumab-treated patients, compared with 9.7 months in docetaxel-treated patients (hazard ratio [HR], 0.73; P = .04) and in the OAK trial, over a median follow-up of 21 months, the median OS was 15.7 months and 10.3 months, respectively.
The main difference between the 2 trials was that the OS benefit in the POPLAR trial reached statistical significance only in patients with the highest levels of PD-L1 expression, whereas in the OAK study, the OS was significantly improved regardless of PD-L1 expression status, although the greatest benefit was derived in patients with higher levels of PD-L1 expression (20.5 months and 8.9 months, respectively). The OS benefit was observed across all other prespecified subgroups, except in patients with EGFR mutation-positivity in the OAK trial. Progression-free survival and overall response rates were similar in the 2 treatment arms in both studies.
An additional 375 patients were enrolled in the OAK trial and included in the safety analyses. In both studies, the safety profile of atezolizumab was similar to that observed in previous studies of this drug. The most commonly observed adverse events (AEs) with atezolizumab treatment were fatigue, decreased appetite, dyspnea, cough, nausea, musculoskeletal pain, and constipation. Despite longer treatment duration, there were fewer incidences of grade 3/4 AEs with atezolizumab compared with docetaxel (37% vs 54%, respectively, in the OAK trial), a lower rate of discontinuation with atezolizumab treatment, and no deaths related to the study drug. The most common grade 3/4 AEs included dyspnea, pneumonia, hypoxia, hyponatremia, fatigue, and anemia. Clinically significant immune-related AEs included pneumonitis, hepatitis, colitis, and thyroid disease.
Atezolizumab is marketed as Tecentriq by Genentech and the prescribing information details warnings and precautions about immune-related AEs; pneumonitis, colitis, endocrinopathies (eg, thyroid disorders, adrenal insufficiency, and diabetes mellitus), and others, as well as infections, infusion-related reactions, and embryofetal toxicity.3
Atezolizumab treatment should be withheld for grade 2 pneumonitis; aspartate aminotransferase (AST) or alanine aminotransferase (ALT) levels of >3-5 times the upper limit of normal (ULN) or total bilirubin levels of >1.5 and up to 3 times the ULN; grade 2/3 diarrhea or colitis, adrenal insufficiency, hypothyroidism, or grade 3/4 hyperglycemia; grade 2 ocular inflammatory toxicity; grade 2/3 pancreatitis or grade 3/4 increases in amylase or lipase levels; grade 3/4 infection; grade 2 infusion-related reactions; and grade 3 rash. Treatment can then be resumed in patients whose AEs recover to grade 0 or 1.
Treatment should be permanently discontinued in the event of grade 3/4 pneumonitis; AST/ALT levels of >5 times the ULN or bilirubin levels of >3 times the ULN; grade 4 diarrhea or colitis; myasthenic syndrome/myasthenia gravis, Guillain-Barre syndrome or meningoencephalitis (all grades); grade 3/4 ocular inflammatory toxicity; grade 4 or recurrent (any grade) pancreatitis; grade 3/4 infusion-related reactions; or grade 4 rash. Patients should also be advised of the risk of fetal harm.
The approval last fall by the US Food and Drug Administration (FDA) of the immune checkpoint inhibitor atezolizumab for the treatment of metastatic non–small cell lung cancer (NSCLC) marked the second approved indication for the drug in a single year. Atezolizumab, which targets the programmed cell death protein-ligand 1 (PD-L1), has a unique mechanism of action compared with other approved immune checkpoint inhibitors and had been previously approved for the treatment of patients with advanced urothelial carcinoma. The current approval was based on 2 international, randomized, open-label trials, involving more than 1,000 patients, in which atezolizumab outperformed the chemotherapeutic drug docetaxel.
The POPLAR trial1 was a phase 2 study performed at 61 academic centers and community oncology practices across 13 countries in Europe and North America and enrolled 287 patients, while the phase 3 OAK trial2 was carried out at 193 centers in 31 countries and enrolled 850 patients.
Eligibility criteria for the 2 studies were similar; patients were 18 years or older, with an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, had measurable disease as per Response Evaluation Criteria in Solid Tumors (RECIST, version 1.1), and adequate hematologic and end-organ function. Patients with asymptomatic or treated central nervous system (CNS) metastases were also eligible.
Patients were excluded from both studies if they had a history of autoimmune disease, pneumonitis or chronic viral diseases, or had received previous treatment with docetaxel, CD137 agonists, anti-CTLA4, anti-PD-L1 or anti-PD-1 therapeutics.
Patients were randomized 1:1 to receive atezolizumab, administered intravenously at a fixed dose of 1,200 mg, or a 75 mg/m2 dose of docetaxel, every 3 weeks on day 1 of each 3-week cycle. Randomization was stratified according to PD-L1 expression, number of previous chemotherapy regimens, and histology (squamous vs nonsquamous). Tumors were assessed at baseline and every 6 weeks for 36 weeks, and every 9 weeks thereafter until progression or, in patients who continued beyond progression, until discontinuation. PD-L1 expression was assessed prospectively on tumor cells and tumor-infiltrating immune cells, using the FDA-approved companion diagnostic, Ventana SP142 PD-L1 immunohistochemistry assay.
The primary endpoint in both studies was overall survival (OS), which was significantly improved in the atezolizumab treatment arm. In the POPLAR trial, over a minimum follow-up of 13 months, the median OS was 12.6 months in atezolizumab-treated patients, compared with 9.7 months in docetaxel-treated patients (hazard ratio [HR], 0.73; P = .04) and in the OAK trial, over a median follow-up of 21 months, the median OS was 15.7 months and 10.3 months, respectively.
The main difference between the 2 trials was that the OS benefit in the POPLAR trial reached statistical significance only in patients with the highest levels of PD-L1 expression, whereas in the OAK study, the OS was significantly improved regardless of PD-L1 expression status, although the greatest benefit was derived in patients with higher levels of PD-L1 expression (20.5 months and 8.9 months, respectively). The OS benefit was observed across all other prespecified subgroups, except in patients with EGFR mutation-positivity in the OAK trial. Progression-free survival and overall response rates were similar in the 2 treatment arms in both studies.
An additional 375 patients were enrolled in the OAK trial and included in the safety analyses. In both studies, the safety profile of atezolizumab was similar to that observed in previous studies of this drug. The most commonly observed adverse events (AEs) with atezolizumab treatment were fatigue, decreased appetite, dyspnea, cough, nausea, musculoskeletal pain, and constipation. Despite longer treatment duration, there were fewer incidences of grade 3/4 AEs with atezolizumab compared with docetaxel (37% vs 54%, respectively, in the OAK trial), a lower rate of discontinuation with atezolizumab treatment, and no deaths related to the study drug. The most common grade 3/4 AEs included dyspnea, pneumonia, hypoxia, hyponatremia, fatigue, and anemia. Clinically significant immune-related AEs included pneumonitis, hepatitis, colitis, and thyroid disease.
Atezolizumab is marketed as Tecentriq by Genentech and the prescribing information details warnings and precautions about immune-related AEs; pneumonitis, colitis, endocrinopathies (eg, thyroid disorders, adrenal insufficiency, and diabetes mellitus), and others, as well as infections, infusion-related reactions, and embryofetal toxicity.3
Atezolizumab treatment should be withheld for grade 2 pneumonitis; aspartate aminotransferase (AST) or alanine aminotransferase (ALT) levels of >3-5 times the upper limit of normal (ULN) or total bilirubin levels of >1.5 and up to 3 times the ULN; grade 2/3 diarrhea or colitis, adrenal insufficiency, hypothyroidism, or grade 3/4 hyperglycemia; grade 2 ocular inflammatory toxicity; grade 2/3 pancreatitis or grade 3/4 increases in amylase or lipase levels; grade 3/4 infection; grade 2 infusion-related reactions; and grade 3 rash. Treatment can then be resumed in patients whose AEs recover to grade 0 or 1.
Treatment should be permanently discontinued in the event of grade 3/4 pneumonitis; AST/ALT levels of >5 times the ULN or bilirubin levels of >3 times the ULN; grade 4 diarrhea or colitis; myasthenic syndrome/myasthenia gravis, Guillain-Barre syndrome or meningoencephalitis (all grades); grade 3/4 ocular inflammatory toxicity; grade 4 or recurrent (any grade) pancreatitis; grade 3/4 infusion-related reactions; or grade 4 rash. Patients should also be advised of the risk of fetal harm.